Advanced Palladium-Catalyzed Asymmetric Synthesis of Chiral 3,4-Dihydroisoquinolinone Intermediates for Scalable Production
Introduction to Patent CN110204491B: A Breakthrough in Chiral Scaffold Construction
The pharmaceutical industry continuously demands efficient access to complex chiral heterocycles, particularly the 3,4-dihydroisoquinolinone skeleton which serves as a privileged structure in numerous bioactive natural products and drug candidates. Patent CN110204491B, published in early 2021, discloses a highly innovative synthetic methodology that addresses long-standing challenges in constructing these motifs. The invention details a palladium-catalyzed asymmetric C-H functionalization tandem reaction between N-benzoyl sulfonamide compounds and conjugated dienes. This approach represents a significant paradigm shift from traditional multi-step syntheses, offering a direct, atom-economical route to high-value chiral intermediates. By leveraging a carefully tuned catalytic system involving specific chiral ligands and mild oxidants, the process achieves remarkable levels of regioselectivity and enantioselectivity. For R&D directors and process chemists, this technology opens new avenues for the rapid assembly of diverse libraries of isoquinoline derivatives, potentially accelerating lead optimization campaigns in medicinal chemistry programs focused on neurological or oncological targets.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of chiral 3,4-dihydroisoquinolinone cores has relied heavily on classical named reactions such as the Bischler-Napieralski cyclization, Pomeranz-Fritsch-Bobbitt reaction, or Friedel-Crafts acylation. These traditional pathways are fraught with significant operational and economic drawbacks that hinder large-scale manufacturing. Primarily, they often necessitate the use of pre-functionalized chiral amines as starting materials, which are frequently expensive, commercially scarce, or require resolution from racemic mixtures, thereby capping the maximum theoretical yield at 50%. Furthermore, these methods typically involve harsh reaction conditions, including strong acids or Lewis acids at elevated temperatures, which can lead to poor functional group tolerance and the generation of substantial hazardous waste. More recent attempts utilizing rhodium-cyclopentadienyl catalysts for [4+2] cycloadditions have shown promise but are limited by the prohibitive cost and synthetic complexity of the rhodium catalysts themselves. Additionally, earlier palladium-catalyzed attempts often suffered from low yields, narrow substrate scope, or a lack of stereocontrol, frequently producing unwanted five-membered rings via aza-Wacker-type pathways instead of the desired six-membered lactams.
The Novel Approach
The methodology outlined in CN110204491B overcomes these historical bottlenecks through a sophisticated asymmetric C-H activation strategy. By employing an N-sulfonylbenzamide directing group, the process successfully guides the palladium catalyst to activate specific sp2 C-H bonds, facilitating a tandem cyclization with 1,3-conjugated dienes. This direct functionalization eliminates the need for pre-halogenated substrates or chiral amine precursors, drastically simplifying the synthetic sequence. The reaction proceeds under relatively mild conditions (80°C) in an air atmosphere, utilizing trifluorotoluene as a solvent, which enhances the safety profile and operational ease compared to strictly anaerobic protocols. Crucially, the system suppresses competitive beta-hydride elimination pathways that typically plague palladium-olefin interactions, ensuring high selectivity for the six-membered ring formation. As illustrated in the reaction scheme below, the transformation converts simple, achiral starting materials directly into complex chiral architectures with high fidelity.
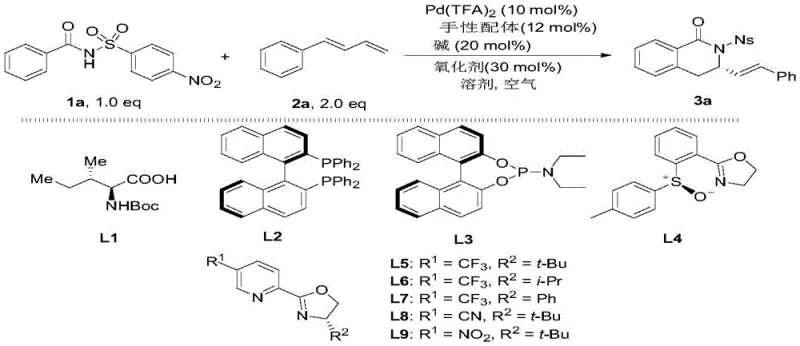
This novel approach not only streamlines the synthesis but also expands the chemical space accessible to medicinal chemists, allowing for the late-stage introduction of diverse substituents on both the aromatic ring and the styryl side chain without compromising stereochemical integrity.
Mechanistic Insights into Pd-Catalyzed Asymmetric C-H Functionalization
The success of this transformation hinges on a delicate balance of electronic and steric factors within the catalytic cycle. The mechanism initiates with the coordination of the palladium(II) species to the nitrogen atom of the N-benzoyl sulfonamide, acting as a bidentate directing group. This coordination facilitates the concerted metalation-deprotonation (CMD) of the ortho C-H bond, generating a stable five-membered palladacycle intermediate. The presence of the sulfonyl group is critical here, as it modulates the electron density of the nitrogen, enhancing its coordinating ability while preventing unwanted side reactions. Following C-H activation, the conjugated diene coordinates to the palladium center and undergoes migratory insertion. The choice of the chiral ligand, specifically the binaphthyl-derived phosphine L5, creates a chiral pocket that dictates the facial selectivity of this insertion step, thereby establishing the stereogenic center at the C3 position of the dihydroisoquinolinone ring. Subsequent intramolecular nucleophilic attack by the nitrogen onto the carbonyl carbon, followed by reductive elimination or protonolysis, releases the final product and regenerates the active Pd(0) species, which is re-oxidized to Pd(II) by the benzoquinone oxidant to close the catalytic cycle. This intricate interplay ensures that the thermodynamic preference for beta-hydride elimination is overridden by the kinetic favorability of the cyclization pathway.
From an impurity control perspective, the mechanism inherently minimizes the formation of regioisomers. The rigid geometry of the palladacycle intermediate restricts the orientation of the incoming diene, ensuring that insertion occurs exclusively at the terminal position to yield the linear styryl product rather than branched isomers. Furthermore, the use of 2,6-dimethoxy-1,4-benzoquinone as the oxidant is pivotal; it is sufficiently mild to prevent over-oxidation of the sensitive olefinic moiety in the product yet potent enough to sustain the catalytic turnover. The suppression of five-membered ring byproducts, a common issue in similar Pd-catalyzed reactions, is attributed to the specific bite angle and steric bulk of ligand L5, which destabilizes the transition state leading to the smaller ring size. This level of mechanistic control translates directly to a cleaner crude reaction profile, reducing the burden on downstream purification processes and enhancing the overall mass balance of the manufacturing operation.
How to Synthesize Chiral 3,4-Dihydroisoquinolinone Efficiently
Implementing this synthesis requires precise adherence to the optimized parameters identified in the patent examples to ensure reproducibility and high enantiomeric excess. The protocol involves a straightforward one-pot procedure where the N-benzoyl sulfonamide and conjugated diene are combined with the palladium catalyst system in trifluorotoluene. The reaction is heated to 80°C and stirred for 48 hours under an air atmosphere, eliminating the need for complex inert gas manifolds typically associated with sensitive organometallic chemistry. Detailed standardized synthesis steps for this specific transformation are provided in the guide below.
- Charge a reaction vessel with N-benzoyl sulfonamide substrate, palladium trifluoroacetate catalyst, chiral phosphine ligand L5, oxidant, and base in trifluorotoluene solvent.
- Add the conjugated diene reactant and stir the mixture at 80°C under an air atmosphere for approximately 48 hours to facilitate the asymmetric tandem cyclization.
- Upon completion, cool the reaction system to room temperature and purify the crude mixture via column chromatography to isolate the high-purity chiral product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this technology offers tangible strategic benefits beyond mere technical novelty. The shift from precious rhodium catalysts to a palladium-based system represents a significant opportunity for cost reduction in pharmaceutical intermediate manufacturing. Palladium is generally more abundant and less volatile in price compared to specialized rhodium complexes, and the ligand L5, while chiral, is derived from commercially available binaphthyl backbones, ensuring a stable supply chain. Moreover, the use of simple, achiral starting materials like N-benzoyl sulfonamides and commodity dienes removes the dependency on expensive chiral pool resources or resolution processes, further driving down the cost of goods sold (COGS). The reaction's tolerance to air and its operation in a single solvent system simplify the equipment requirements, allowing for execution in standard glass-lined or stainless steel reactors without the need for specialized high-pressure or strictly anaerobic setups.
- Cost Reduction in Manufacturing: The elimination of expensive rhodium catalysts and chiral amine starting materials drastically lowers the raw material expenditure. By utilizing a tandem reaction that builds complexity in a single step, the process reduces the number of unit operations, solvent swaps, and isolation steps required compared to classical multi-step routes. This consolidation of steps leads to substantial savings in labor, energy consumption, and solvent waste disposal costs, making the overall production economics highly favorable for large-scale campaigns.
- Enhanced Supply Chain Reliability: The reliance on broadly available chemical building blocks mitigates the risk of supply disruptions often associated with niche chiral reagents. The robustness of the catalytic system, which functions effectively under air atmosphere, reduces the logistical complexity of transporting and storing sensitive reagents under inert conditions. This operational resilience ensures consistent batch-to-batch quality and reliable delivery schedules, which are critical metrics for maintaining uninterrupted API production lines and meeting regulatory filing timelines.
- Scalability and Environmental Compliance: The high atom economy of this C-H functionalization approach aligns well with green chemistry principles, minimizing the generation of stoichiometric salt waste typical of traditional cyclization methods. The ability to achieve high yields and selectivity reduces the need for extensive chromatographic purification, facilitating a switch to crystallization-based workups which are more scalable and environmentally benign. This efficiency not only lowers the environmental footprint but also accelerates the timeline for technology transfer from laboratory to pilot and commercial plant scales.
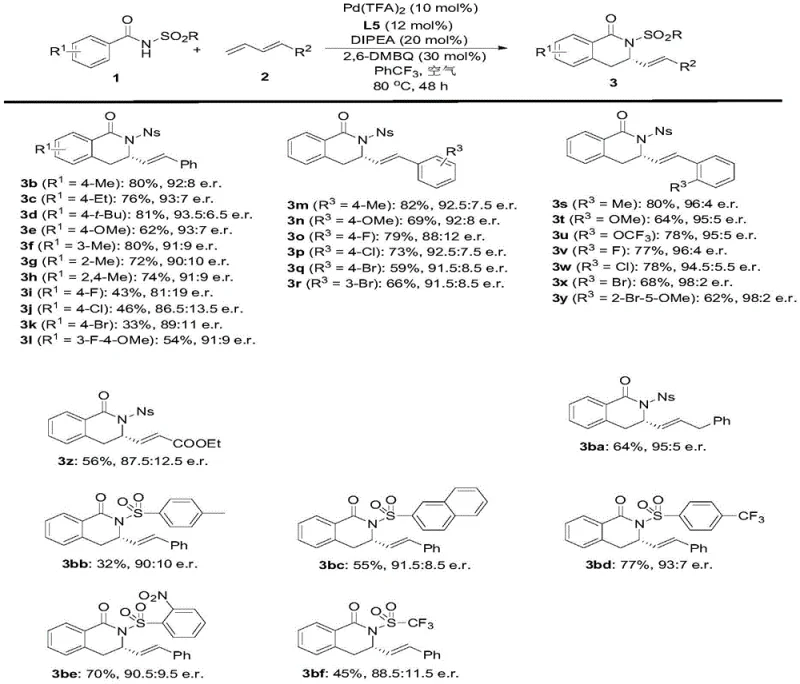
The broad substrate scope demonstrated in the patent, accommodating various electron-donating and withdrawing groups as well as different diene substitutions, confirms the versatility of this platform for generating diverse analog libraries without re-optimizing the core process.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and optimization of this synthetic route, derived directly from the experimental data and comparative analysis presented in the patent documentation. Understanding these nuances is essential for process development teams evaluating the feasibility of this technology for their specific pipeline candidates.
Q: Why is this palladium-catalyzed method preferred over traditional rhodium-catalyzed routes?
A: Traditional methods often rely on expensive and difficult-to-synthesize rhodium-cyclopentadienyl catalysts. This patent utilizes a more economically viable palladium system with readily available ligands, significantly reducing raw material costs while maintaining high enantioselectivity.
Q: What are the key advantages regarding atom economy and step count?
A: Unlike classical Bischler-Napieralski or Pomeranz-Fritsch reactions which require multiple steps and harsh reagents, this asymmetric C-H functionalization tandem reaction constructs the core skeleton directly from simple N-benzoyl sulfonamides and dienes, improving atom economy and operational simplicity.
Q: What represents the optimal catalytic system for this transformation?
A: The patent identifies Pd(TFA)2 combined with the chiral ligand L5, using 2,6-dimethoxy-1,4-benzoquinone as the oxidant and DIPEA as the base in trifluorotoluene, yielding products with up to 98:2 enantiomeric ratios.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral 3,4-Dihydroisoquinolinone Supplier
The technological advancements detailed in CN110204491B underscore the evolving landscape of chiral intermediate synthesis, where efficiency and selectivity are paramount. NINGBO INNO PHARMCHEM stands at the forefront of this evolution, possessing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facility is equipped with rigorous QC labs and stringent purity specifications to ensure that every batch of chiral 3,4-dihydroisoquinolinone meets the exacting standards required for global pharmaceutical registration. We understand the critical nature of supply continuity and are committed to delivering high-purity intermediates that enable our partners to advance their drug discovery programs with confidence.
We invite you to engage with our technical procurement team to discuss how this novel palladium-catalyzed route can be integrated into your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the potential economic benefits specific to your target molecule. We encourage you to contact us today to obtain specific COA data and route feasibility assessments tailored to your project needs, ensuring a seamless transition from bench-scale innovation to commercial reality.