Revolutionizing Pharmaceutical Intermediate Synthesis with Direct C-H Alkenylation Technology
Revolutionizing Pharmaceutical Intermediate Synthesis with Direct C-H Alkenylation Technology
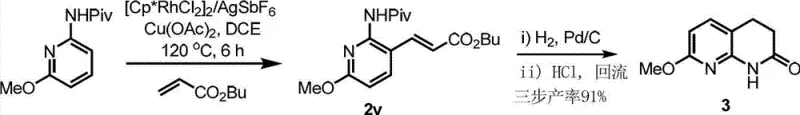
The pharmaceutical industry continuously seeks robust synthetic methodologies that can deliver complex heterocyclic scaffolds with high efficiency and minimal environmental impact. A pivotal advancement in this domain is detailed in Chinese Patent CN103113291A, which discloses a novel synthesis method for 3-position alkenylpyridine derivatives. These compounds serve as critical precursors for substituted 3,4-dihydro-1,8-naphthyridinones, a privileged structural motif found in numerous bioactive molecules ranging from Fab inhibitors to tuberculosis treatments. The significance of this technology lies in its ability to bypass traditional, wasteful functionalization steps, directly installing alkenyl groups onto the pyridine core through a sophisticated transition metal-catalyzed C-H activation process. This breakthrough not only streamlines the synthetic route but also opens new avenues for the rapid diversification of drug candidates, addressing the urgent need for reliable pharmaceutical intermediate suppliers who can offer cost-effective and scalable solutions.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of 3-alkenylpyridine frameworks has relied heavily on classical organic transformations such as the palladium-catalyzed Heck reaction or the Wittig olefination. While these methods are well-established in academic literature, they present substantial hurdles when applied to large-scale commercial manufacturing. The Heck reaction, for instance, typically necessitates the use of pre-halogenated pyridine starting materials, which are often expensive, hazardous to handle, and generate stoichiometric amounts of salt waste during the coupling process. Furthermore, achieving high regioselectivity at the 3-position of the pyridine ring using these conventional tactics is notoriously difficult, often leading to complex mixtures of isomers that require energy-intensive and yield-reducing purification steps. The cumulative effect of these inefficiencies is a significant escalation in the cost of goods sold (COGS) and extended lead times, creating bottlenecks for procurement managers striving to optimize their supply chains for active pharmaceutical ingredients.
The Novel Approach
In stark contrast to these legacy techniques, the methodology outlined in the patent utilizes a direct C-H bond activation strategy mediated by a rhodium catalyst system. This approach fundamentally redefines the synthetic logic by treating the C-H bond itself as the reactive handle, thereby eliminating the need for prior halogenation. By employing 2-acylaminopyridines as substrates and electron-deficient olefins as coupling partners, the reaction achieves exceptional regioselectivity exclusively at the 3-position of the pyridine ring. The operational simplicity is another major advantage; the reaction proceeds efficiently in common organic solvents like 1,2-dichloroethane under heating conditions, without the stringent requirement for anhydrous or oxygen-free atmospheres that often complicate glovebox-dependent organometallic chemistry. This robustness translates directly into enhanced process safety and reduced infrastructure costs for manufacturing facilities aiming for cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Rhodium-Catalyzed C-H Activation
The efficacy of this synthesis relies on a meticulously orchestrated catalytic cycle involving a trivalent rhodium species. The mechanism initiates with the coordination of the pyridine nitrogen atom to the metal center, followed by the generation of an active cationic rhodium(III) complex through the interaction with a silver salt additive such as silver hexafluoroantimonate. Under the directive influence of the acyl amino group, the rhodium center selectively activates the proximal C-H bond at the 3-position, forming a stable six-membered metallacycle transition state. This key intermediate then undergoes migratory insertion of the electron-deficient olefin, followed by beta-hydride elimination to release the alkenylated product. Crucially, the catalytic turnover is sustained by a copper(II) oxidant, which re-oxidizes the reduced rhodium(I) species back to the active rhodium(III) state, ensuring the cycle continues with high turnover numbers. Understanding this mechanistic pathway is vital for R&D directors as it highlights the precise control over impurity profiles and the potential for further optimization of ligand environments.
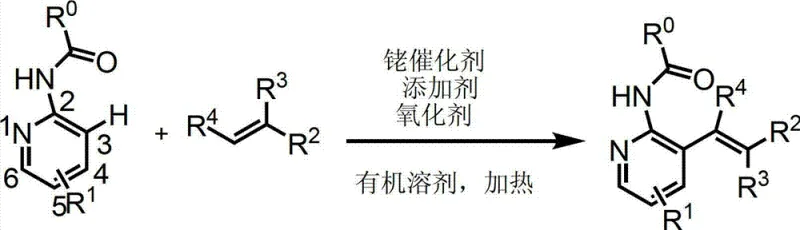
From a quality control perspective, the mechanism inherently favors the formation of the desired trans-alkenylated product due to the stereoelectronic requirements of the beta-hydride elimination step. The use of a pivaloyl (Piv) protecting group on the nitrogen atom is particularly strategic, as its steric bulk and strong coordinating ability enhance both the reaction rate and the selectivity, minimizing the formation of undesired side products or oligomers. This high level of chemoselectivity means that the crude reaction mixture is significantly cleaner compared to traditional cross-coupling reactions, reducing the burden on downstream purification units. For technical teams, this implies that rigorous QC labs can achieve stringent purity specifications more consistently, ensuring that the final API intermediates meet the demanding regulatory standards required for global pharmaceutical markets without excessive reprocessing.
How to Synthesize 3-Position Alkenylpyridine Derivatives Efficiently
Implementing this technology requires careful attention to reagent stoichiometry and reaction parameters to maximize yield and throughput. The patent provides a clear blueprint where 2-pivalamidopyridine reacts with an excess of electron-deficient olefin in the presence of a rhodium dimer catalyst and silver additive. The process is designed to be operationally straightforward, utilizing standard heating equipment rather than specialized high-pressure reactors, which facilitates easier technology transfer from laboratory to pilot plant scales. Detailed standardized synthesis steps are provided below to guide process chemists in replicating these high-efficiency results.
- Combine 2-pivalamidopyridine, electron-deficient olefin, rhodium catalyst, silver additive, and copper oxidant in 1,2-dichloroethane solvent.
- Heat the reaction mixture to 110-130°C in a sealed tube for approximately 24 hours to ensure complete conversion.
- Purify the resulting crude mixture via silica gel column chromatography to isolate the high-purity 3-alkenylpyridine derivative.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this rhodium-catalyzed C-H activation technology represents a strategic opportunity to de-risk the supply of critical heterocyclic building blocks. By shifting away from halogenated starting materials, manufacturers can mitigate the volatility associated with the pricing and availability of halogenated pyridines, which are often subject to fluctuating market dynamics and environmental regulations regarding halogen waste disposal. The simplified synthetic route reduces the total number of unit operations, directly correlating to lower utility consumption and reduced labor hours per kilogram of product. This streamlined process flow enhances overall equipment effectiveness (OEE) and allows for greater flexibility in production scheduling, ensuring that delivery commitments to downstream API manufacturers are met with greater reliability and consistency.
- Cost Reduction in Manufacturing: The elimination of the pre-halogenation step removes a significant cost driver from the bill of materials, as halogenated heterocycles are typically more expensive than their non-halogenated counterparts. Furthermore, the high atom economy of the direct C-H coupling minimizes waste generation, leading to substantial cost savings in waste treatment and disposal fees. The use of relatively inexpensive oxidants like copper acetate instead of costly noble metal oxidants further optimizes the variable costs associated with large-scale production, driving down the overall manufacturing expense without compromising on product quality.
- Enhanced Supply Chain Reliability: The raw materials required for this synthesis, including 2-acylaminopyridines and common acrylates, are widely available from multiple global chemical suppliers, reducing the risk of single-source dependency. The robustness of the reaction conditions, which do not demand extreme temperatures or pressures, ensures that production can be maintained across different manufacturing sites with minimal variance in output quality. This geographical flexibility strengthens the supply chain resilience, allowing for diversified sourcing strategies that protect against regional disruptions and ensure continuous availability of high-purity pharmaceutical intermediates for critical drug development programs.
- Scalability and Environmental Compliance: The process demonstrates excellent scalability potential, as evidenced by the successful execution of reactions in sealed tubes at moderate temperatures, which can be readily adapted to standard stirred-tank reactors. The avoidance of toxic halogenated byproducts aligns with increasingly stringent environmental, health, and safety (EHS) regulations, simplifying the permitting process for new manufacturing lines. This green chemistry profile not only reduces the environmental footprint but also enhances the corporate sustainability metrics of the supply chain, a factor that is becoming progressively important for multinational pharmaceutical companies evaluating their vendor partnerships.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing a transparent view of the method's capabilities and limitations for potential partners.
Q: What are the primary limitations of conventional pyridine alkenylation methods?
A: Traditional methods such as palladium-catalyzed Heck reactions or Wittig condensations often suffer from low yields, poor functional group tolerance, and require cumbersome multi-step sequences involving pre-halogenated substrates.
Q: How does the rhodium-catalyzed method improve regioselectivity?
A: The use of a pivaloyl directing group combined with a trivalent rhodium catalyst ensures highly selective activation of the C-H bond specifically at the 3-position of the pyridine ring, simplifying downstream purification.
Q: Is this synthesis method scalable for commercial production?
A: Yes, the process operates under relatively mild heating conditions (110-130°C) without requiring strict anhydrous or oxygen-free environments, making it highly suitable for scale-up in standard industrial reactors.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Alkenylpyridine Derivatives Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced C-H activation technologies in accelerating drug discovery and development timelines. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that innovative laboratory methods like the one described in CN103113291A can be successfully translated into robust industrial processes. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of 3-alkenylpyridine derivatives delivered meets the highest standards of quality and consistency required by the global pharmaceutical industry.
We invite you to collaborate with our technical procurement team to explore how this efficient synthesis route can be integrated into your specific project requirements. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic benefits tailored to your volume needs. We encourage you to contact us today to obtain specific COA data and comprehensive route feasibility assessments, ensuring that your supply chain is optimized for both performance and profitability in the competitive landscape of API manufacturing.