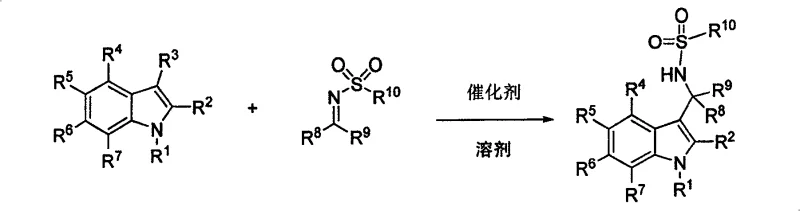
Scalable Synthesis of Optically Active 3-Aminoindole Intermediates via Chiral Phosphoric Acid Catalysis
The pharmaceutical and fine chemical industries are constantly seeking more efficient, sustainable, and compliant pathways for synthesizing complex chiral building blocks. Patent CN100548979C introduces a groundbreaking methodology for the synthesis of 3-methylaminoindole compounds, a structural motif prevalent in numerous biologically active alkaloids and drug candidates. This technology leverages the power of chiral phosphoric acid organocatalysis to drive the asymmetric Friedel-Crafts reaction between indoles and sulfonimides. Unlike traditional approaches that rely heavily on transition metals, this novel process operates under mild conditions, delivering high yields ranging from 60% to 98% and exceptional enantioselectivity exceeding 99% ee in optimized cases. For R&D directors and procurement specialists, this represents a paradigm shift towards greener, metal-free chemistry that simplifies downstream processing and regulatory approval.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of chiral 3-substituted indoles has been fraught with challenges, primarily revolving around the reliance on transition metal catalysts. Conventional synthetic routes often necessitate the use of expensive and toxic heavy metals, which pose significant environmental hazards and create stringent regulatory hurdles for pharmaceutical manufacturing. The presence of residual metal ions in the final Active Pharmaceutical Ingredient (API) requires costly and time-consuming purification steps to meet International Council for Harmonisation (ICH) guidelines. Furthermore, many traditional methods suffer from poor atom economy, require harsh reaction conditions such as extreme temperatures or pressures, and often exhibit sluggish kinetics, with reaction times extending over several days. These inefficiencies not only inflate the cost of goods sold (COGS) but also introduce variability in batch-to-batch consistency, complicating the commercial scale-up of complex pharmaceutical intermediates.
The Novel Approach
The methodology disclosed in CN100548979C offers a robust alternative by utilizing chiral organic phosphoric acids as Brønsted acid catalysts. This metal-free approach fundamentally alters the reaction landscape, enabling the direct asymmetric addition of various indole derivatives to sulfonimides with remarkable precision. The reaction proceeds efficiently in common organic solvents like toluene or dichloromethane at temperatures ranging from -60°C to 25°C, significantly reducing energy consumption. By eliminating the need for metal salts, the process inherently avoids heavy metal contamination, thereby streamlining the purification workflow and ensuring a cleaner impurity profile. This operational simplicity, combined with the ability to tune the catalyst structure for specific substrates, makes this technology highly adaptable for the reliable production of high-purity pharmaceutical intermediates on a commercial scale.
Mechanistic Insights into Chiral Phosphoric Acid Catalyzed Friedel-Crafts Reaction
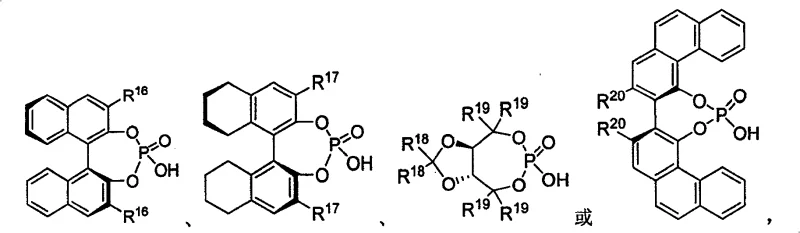
The core of this technological advancement lies in the unique activation mode of the chiral phosphoric acid catalyst. These catalysts, typically derived from BINOL (1,1'-bi-2-naphthol) scaffolds, function as dual hydrogen-bond donors. In the transition state, the phosphoryl oxygen activates the electrophilic sulfonimine substrate via hydrogen bonding, while the acidic proton simultaneously interacts with the nucleophilic indole. This bifunctional activation organizes the reactants within a well-defined chiral pocket created by the bulky substituents at the 3,3'-positions of the binaphthyl backbone. This steric environment dictates the facial selectivity of the indole attack, ensuring the formation of the desired enantiomer with high fidelity. The strength of the hydrogen bonding network can be finely modulated by altering the electronic properties of the aryl groups attached to the phosphorus center, allowing chemists to optimize both reaction rate and stereoselectivity for diverse substrate classes.
From an impurity control perspective, the organocatalytic nature of this reaction provides distinct advantages. Since no transition metals are involved, there is no risk of metal-catalyzed side reactions such as oxidative degradation or unwanted coupling processes that often plague metal-mediated syntheses. The primary byproducts are typically unreacted starting materials or diastereomers, which are generally easier to separate via standard crystallization or chromatography techniques. Furthermore, the mild acidic conditions prevent the decomposition of sensitive functional groups often present on complex indole scaffolds, such as halogens or methoxy groups. This chemoselectivity ensures that the structural integrity of the molecule is maintained throughout the synthesis, resulting in a cleaner crude product and reducing the burden on quality control laboratories during the release testing of the final intermediate.
How to Synthesize 3-Methylaminoindole Efficiently
The practical implementation of this synthesis involves a straightforward protocol suitable for both laboratory and pilot plant settings. The process begins with the preparation of the chiral phosphoric acid catalyst, which can be synthesized from commercially available binaphthol derivatives. In a typical run, the catalyst is dissolved in a dry organic solvent under an inert atmosphere, followed by the addition of the sulfonimine electrophile. After a brief pre-stirring period to establish the catalyst-substrate complex, the indole nucleophile is introduced at controlled low temperatures. The reaction progress is monitored until completion, after which a simple aqueous workup removes the catalyst and salts. Detailed standardized synthesis steps see the guide below.
- Prepare the reaction mixture by dissolving the chiral phosphoric acid catalyst and sulfonimide substrate in an organic solvent such as toluene under inert atmosphere.
- Cool the reaction mixture to the specified temperature range, typically between -60°C and 25°C, to ensure optimal stereocontrol.
- Add the indole compound to the mixture and stir for the designated time, followed by aqueous workup and purification via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this organocatalytic technology translates into tangible strategic benefits beyond mere chemical elegance. The elimination of precious metal catalysts removes a significant cost driver and supply chain bottleneck, as the prices of metals like palladium or rhodium can be volatile and their availability subject to geopolitical constraints. Moreover, the simplified purification process reduces the consumption of silica gel and solvents, leading to substantial cost savings in waste management and raw material usage. The robustness of the reaction conditions ensures high reproducibility, which is critical for maintaining consistent supply to downstream API manufacturers without unexpected delays caused by failed batches or complex rework procedures.
- Cost Reduction in Manufacturing: The removal of expensive transition metal catalysts and the associated scavenging agents drastically lowers the direct material costs of the synthesis. Additionally, the high yields and selectivity minimize the loss of valuable starting materials, improving the overall mass balance of the process. By avoiding the need for specialized equipment to handle pyrophoric metal reagents or extensive filtration systems for metal removal, capital expenditure for production facilities is also optimized, contributing to a more competitive pricing structure for the final intermediate.
- Enhanced Supply Chain Reliability: The starting materials, including various substituted indoles and sulfonimides, are readily available from multiple global suppliers, reducing the risk of single-source dependency. The mild reaction conditions allow for flexible scheduling and do not require extreme cooling or heating infrastructure, making the process easier to integrate into existing multipurpose manufacturing plants. This flexibility enhances the agility of the supply chain, enabling faster response times to market demands and reducing the lead time for high-purity pharmaceutical intermediates required for clinical trial material production.
- Scalability and Environmental Compliance: The metal-free nature of this process aligns perfectly with modern green chemistry principles and increasingly strict environmental regulations. The absence of heavy metal waste simplifies effluent treatment and disposal, lowering the environmental compliance burden. The reaction has been demonstrated to scale effectively from milligram to gram scales in the patent examples, indicating strong potential for ton-scale production. This scalability ensures that the technology can support the entire product lifecycle, from early-stage drug discovery to commercial manufacturing, without the need for disruptive process changes.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this chiral phosphoric acid catalyzed synthesis. These insights are derived directly from the experimental data and scope defined in the patent literature, providing a clear understanding of the technology's capabilities and limitations for potential partners and licensees.
Q: What are the advantages of using chiral phosphoric acid over metal catalysts for this synthesis?
A: Chiral phosphoric acids eliminate the risk of heavy metal contamination, which is critical for pharmaceutical compliance. They also offer milder reaction conditions and often shorter reaction times compared to traditional metal-catalyzed methods.
Q: What is the typical enantiomeric excess (ee) achievable with this method?
A: According to patent CN100548979C, the method achieves high enantioselectivity, generally ranging from 58% to greater than 99% ee, depending on the specific catalyst and substrate combination used.
Q: Can the sulfonyl protecting group be removed after the reaction?
A: Yes, the patent demonstrates that the sulfonyl group can be effectively removed under standard reductive conditions (e.g., Na/NH3) to yield the free amine, which can then be further functionalized with other protecting groups like Cbz.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Aminoindole Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthetic methodologies to stay ahead in the competitive pharmaceutical landscape. Our team of expert chemists has extensively evaluated the organocatalytic routes described in CN100548979C and possesses the technical expertise to adapt and optimize these pathways for your specific project needs. We have extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from lab bench to manufacturing plant is seamless. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of chiral 3-aminoindole intermediate meets the highest industry standards for enantiomeric excess and chemical purity.
We invite you to collaborate with us to leverage this cutting-edge technology for your drug development programs. Whether you require custom synthesis of novel analogs or large-scale production of established intermediates, our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements. Contact us today to request specific COA data and route feasibility assessments, and let us demonstrate how our commitment to innovation and quality can drive value for your organization.