Revolutionizing 3,4-Dihydropyrimidinone Production with Efficient Nucleophilic Dearomatization Technology
The pharmaceutical industry continuously seeks robust synthetic methodologies to access privileged scaffolds with high efficiency and structural diversity. Patent CN113173951B introduces a groundbreaking approach for the synthesis of 3,4-dihydropyrimidinone derivatives through a nucleophilic dearomatization strategy. This technology addresses critical limitations in current heterocyclic chemistry by enabling the direct functionalization of simple 2-hydroxypyrimidines using phosphite esters as nucleophilic reagents. The resulting compounds, which feature a phosphate substituent at the C4 position, represent a valuable class of intermediates with potential applications in anticancer, anti-inflammatory, and calcium channel inhibitor drug development. By leveraging a Lewis acid-catalyzed mechanism, this method operates under remarkably mild conditions, typically between room temperature and 100°C, ensuring high yields and operational simplicity that are essential for modern reliable pharmaceutical intermediate supplier networks.
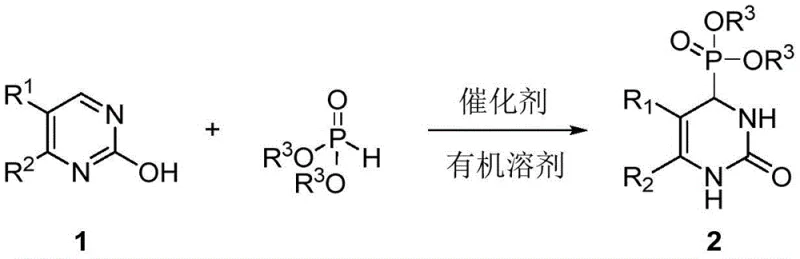
The significance of this invention lies in its ability to bypass the restrictive substrate scope of classical multicomponent reactions. Traditional methods often struggle to introduce heteroatom-based substituents at the chiral C4 center of the dihydropyrimidinone ring, limiting the chemical space available for medicinal chemists. This patent discloses a versatile pathway where various dialkyl phosphites, including dimethyl, diethyl, and diisopropyl phosphites, react efficiently with substituted 2-hydroxypyrimidines. The reaction tolerates a wide range of functional groups on the phenyl ring, such as chloro, fluoro, methyl, and methoxy substituents, demonstrating exceptional compatibility. This broad substrate tolerance is crucial for cost reduction in pharmaceutical intermediate manufacturing, as it allows for the use of diverse, commercially available starting materials without requiring extensive protection-deprotection sequences or harsh reaction environments that degrade sensitive functional groups.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of dihydropyrimidinones has relied heavily on the Biginelli three-component condensation reaction. While useful, this classical approach suffers from significant drawbacks that hinder its utility in advanced drug discovery programs. The primary limitation is the narrow substrate scope, which predominantly allows for the construction of dihydropyrimidinones substituted with aryl or alkyl groups at the 4-position. Attempts to introduce heteroatoms or more complex phosphorus-containing motifs at this critical chiral center often result in poor yields, side reactions, or complete failure of the cyclization. Furthermore, traditional Biginelli reactions frequently require strong acidic conditions, high temperatures, and long reaction times, which can lead to the decomposition of sensitive substrates and the formation of difficult-to-remove impurities. These factors collectively increase the cost of goods and complicate the purification process, making it challenging to achieve the high-purity pharmaceutical intermediates required for clinical trials and commercial production.
The Novel Approach
In stark contrast, the nucleophilic dearomatization method described in patent CN113173951B offers a transformative solution to these synthetic challenges. By utilizing 2-hydroxypyrimidines as the starting material, the reaction exploits the keto-enol tautomerism to reduce aromaticity, thereby facilitating a smooth nucleophilic attack by the phosphite ester. This mechanism allows for the direct installation of a phosphonate group at the C4 position, creating a new chiral center with high stereochemical potential. The reaction proceeds efficiently with catalyst loadings as low as 3 mol%, specifically using Zirconium Tetrachloride (ZrCl4), which acts as a potent Lewis acid to activate the substrate. The mild conditions, often achievable at room temperature in solvents like THF, ensure that the integrity of the molecular framework is maintained. This approach not only expands the chemical diversity of accessible dihydropyrimidinones but also streamlines the synthesis workflow, supporting the commercial scale-up of complex pharmaceutical intermediates with improved safety and environmental profiles.
Mechanistic Insights into ZrCl4-Catalyzed Nucleophilic Dearomatization
The core of this synthetic breakthrough relies on the precise activation of the 2-hydroxypyrimidine ring by the zirconium catalyst. ZrCl4 functions as a strong Lewis acid, coordinating with the nitrogen and oxygen atoms of the pyrimidine ring to enhance its electrophilicity. This coordination stabilizes the transition state and lowers the activation energy required for the nucleophilic attack. Simultaneously, the existence of the keto tautomer of the 2-hydroxypyrimidine reduces the aromatic stabilization energy of the ring, making it susceptible to dearomatization. The phosphite ester, acting as a soft nucleophile, attacks the activated C4 position, leading to the formation of a tetrahedral intermediate. Subsequent proton transfer and rearomatization steps, driven by the thermodynamic stability of the carbonyl group in the product, yield the final 3,4-dihydropyrimidinone structure. This mechanistic pathway is highly efficient, minimizing side reactions and ensuring that the phosphorus moiety is incorporated cleanly into the heterocyclic skeleton.
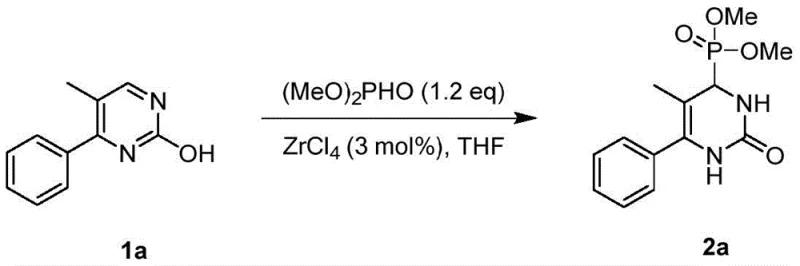
Impurity control is another critical aspect where this mechanism excels. The high selectivity of the ZrCl4-catalyzed reaction minimizes the formation of by-products commonly associated with acid-catalyzed condensations, such as polymeric tars or elimination products. The reaction conditions are sufficiently mild to prevent the hydrolysis of the phosphonate ester or the degradation of sensitive substituents on the phenyl ring. Experimental data shows that yields can reach up to 99% for certain substrates, indicating near-quantitative conversion with minimal waste. This high level of purity simplifies downstream processing, as the crude reaction mixture often requires only standard column chromatography to isolate the target compound. For R&D teams, this means faster iteration cycles and more reliable data during biological screening, as the compounds tested are free from confounding impurities that could skew assay results.
How to Synthesize 3,4-Dihydropyrimidinone Efficiently
The practical implementation of this synthesis route is designed for ease of execution in both laboratory and pilot plant settings. The procedure involves simply mixing the 2-hydroxypyrimidine substrate, the dialkyl phosphite reagent, and the ZrCl4 catalyst in a suitable organic solvent such as tetrahydrofuran. The reaction mixture is then stirred at ambient temperature or slightly elevated temperatures for a period ranging from 8 to 24 hours. Upon completion, the target 3,4-dihydropyrimidinone can be isolated through direct purification techniques. The detailed standardized synthesis steps for replicating this high-yielding transformation are outlined in the guide below.
- Prepare the reaction mixture by combining 2-hydroxypyrimidine substrate and dialkyl phosphite in an organic solvent such as THF.
- Add the Lewis acid catalyst, preferably Zirconium Tetrachloride (ZrCl4), at a loading of 0.1 to 10 mol% relative to the substrate.
- Stir the reaction at room temperature to 100°C for 8 to 24 hours, followed by purification via column chromatography to isolate the target dihydropyrimidinone.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this technology offers substantial strategic benefits that align with the goals of cost efficiency and supply reliability. The adoption of this nucleophilic dearomatization route eliminates the need for expensive transition metal catalysts like palladium, which are often required in alternative hydrogenation or coupling strategies. Instead, it utilizes zirconium tetrachloride, a widely available and cost-effective Lewis acid that can be used in very low catalytic amounts. This shift significantly reduces the raw material costs associated with catalyst procurement and removes the regulatory burden of controlling heavy metal residues in the final active pharmaceutical ingredient. Furthermore, the mild reaction conditions translate to lower energy consumption during manufacturing, as there is no need for extreme heating or cryogenic cooling, contributing to a greener and more sustainable production footprint.
- Cost Reduction in Manufacturing: The economic viability of this process is enhanced by the high atom economy and the use of inexpensive, commodity-grade starting materials. The ability to achieve near-quantitative yields reduces the amount of raw material wasted per batch, directly lowering the cost of goods sold. Additionally, the simplified workup procedure minimizes the consumption of solvents and silica gel during purification, further driving down operational expenses. By avoiding complex multi-step sequences and protecting group manipulations, the overall process mass intensity is improved, making it a highly attractive option for large-scale cost reduction in pharmaceutical intermediate manufacturing.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the reliance on readily available chemical building blocks. Both 2-hydroxypyrimidines and dialkyl phosphites are commercially sourced from multiple vendors, reducing the risk of single-source dependency. The robustness of the reaction conditions ensures consistent batch-to-batch quality, which is critical for maintaining uninterrupted supply to downstream drug manufacturers. The short reaction times and straightforward processing also allow for faster turnaround times, enabling suppliers to respond more agilely to fluctuations in market demand and reducing lead time for high-purity pharmaceutical intermediates.
- Scalability and Environmental Compliance: The scalability of this process is supported by its compatibility with standard reactor equipment and common organic solvents. The absence of hazardous reagents like hydrogen gas or strong mineral acids simplifies safety protocols and waste management. The green chemistry attributes, including high yields and mild conditions, facilitate compliance with increasingly stringent environmental regulations. This makes the technology future-proof for long-term commercial production, ensuring that supply chains remain stable and compliant as global sustainability standards evolve.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These answers are derived directly from the experimental data and technical specifications provided in the patent documentation, ensuring accuracy and relevance for industry professionals. Understanding these details is essential for evaluating the feasibility of integrating this route into existing manufacturing pipelines.
Q: What are the primary advantages of this nucleophilic dearomatization method over traditional Biginelli reactions?
A: Unlike the traditional Biginelli reaction which is often limited to constructing 4-position aryl or alkyl substituted dihydropyrimidinones, this novel method utilizes nucleophilic dearomatization to introduce phosphate substituents at the C4 position. This significantly expands the structural diversity available for drug discovery, allowing for the creation of chiral centers with heteroatom substitutions that were previously difficult to access.
Q: What catalyst system provides the optimal yield for this transformation?
A: Experimental data indicates that Zirconium Tetrachloride (ZrCl4) serves as the superior catalyst for this transformation. When used at a loading of 3 mol% in tetrahydrofuran (THF) at room temperature, it achieves yields as high as 98-99%, outperforming other tested catalysts such as TsOH, CuSO4, or CoSO4 which resulted in significantly lower conversions.
Q: Is this process scalable for industrial manufacturing of pharmaceutical intermediates?
A: Yes, the process is highly amenable to scale-up due to its mild reaction conditions ranging from room temperature to 100°C and the use of conventional organic solvents. The simplicity of the workup procedure, which involves direct column chromatography after stirring, combined with the high atom economy and complete conversion of raw materials, supports robust commercial scale-up for complex pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3,4-Dihydropyrimidinone Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of this nucleophilic dearomatization technology for the development of next-generation therapeutics. As a leading CDMO partner, we possess the technical expertise and infrastructure to translate this innovative laboratory method into a robust commercial process. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and consistency. We are committed to delivering products that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation to verify every batch.
We invite you to collaborate with us to leverage this advanced synthetic route for your drug development programs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific project requirements. Please contact us to request specific COA data and route feasibility assessments, and let us demonstrate how our capabilities can accelerate your timeline to market while optimizing your manufacturing costs.