Advanced Metal-Free Synthesis of 4-Aminoquinoline Compounds for Commercial Scale-Up
Introduction to Novel Electrophilic Activation Strategy
The pharmaceutical industry continuously seeks robust synthetic pathways for heterocyclic scaffolds, particularly those with proven biological activity such as the quinoline core. Patent CN113135852B introduces a groundbreaking preparation method for 4-aminoquinoline compounds, utilizing a sophisticated electrophilic activation strategy that fundamentally shifts the paradigm from traditional metal-catalyzed processes. This technology leverages the synergistic interaction between trifluoromethanesulfonic anhydride and triarylphosphine oxide to drive the intramolecular cyclization of beta-arylamino acrylamide precursors. For R&D directors and process chemists, this represents a significant advancement in accessing polysubstituted 4-aminoquinoline derivatives, which are critical intermediates in the development of next-generation antimalarial agents and other therapeutic areas. The elimination of transition metals not only addresses environmental concerns but also streamlines the downstream purification workflow, offering a compelling value proposition for commercial manufacturing.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 4-aminoquinoline derivatives has relied heavily on methodologies that present substantial operational and economic challenges for large-scale production. Traditional routes often necessitate the use of transition metal catalysts, which introduce complex impurity profiles related to heavy metal residues that are strictly regulated in pharmaceutical ingredients. Furthermore, many existing protocols require harsh reaction conditions, including elevated temperatures and prolonged reaction times, which can lead to thermal degradation of sensitive functional groups and reduced overall yields. The reliance on specialized catalysts also creates supply chain vulnerabilities, as the availability and cost of these metals can fluctuate wildly. Additionally, conventional methods frequently suffer from narrow substrate scope, limiting the ability of medicinal chemists to explore diverse chemical space for structure-activity relationship studies without redesigning the entire synthetic route.
The Novel Approach
In stark contrast, the methodology disclosed in CN113135852B offers a metal-free alternative that operates under remarkably mild conditions, typically ranging from -15°C to 40°C. By employing an electrophilic activating reagent system composed of trifluoromethanesulfonic anhydride and triarylphosphine oxide, the process achieves efficient cyclization without the need for external heating or toxic metal species. This approach significantly broadens the functional group tolerance, allowing for the incorporation of diverse substituents such as halogens, alkyl groups, and electron-withdrawing moieties without compromising reaction efficiency. The operational simplicity is further enhanced by the use of common organic solvents like dichloromethane or chloroform, making the transition from laboratory bench to pilot plant seamless. This novel strategy effectively decouples the synthesis complexity from the cost of goods, providing a scalable solution for producing high-purity pharmaceutical intermediates.
Mechanistic Insights into Electrophilic Activation Cyclization
The core innovation of this technology lies in the precise mechanistic pathway facilitated by the electrophilic activating reagent. The reaction initiates with the activation of the carbonyl or amide functionality within the beta-arylamino acrylamide precursor by the trifluoromethanesulfonic anhydride, generating a highly reactive electrophilic intermediate. This activated species is stabilized and modulated by the presence of the triarylphosphine oxide, which acts as a crucial promoter to lower the activation energy barrier for the subsequent intramolecular nucleophilic attack. The aromatic ring then undergoes cyclization to form the quinoline core, followed by aromatization to yield the final 4-aminoquinoline structure. This mechanism avoids the formation of radical species often associated with metal catalysis, thereby reducing the generation of side products and oligomers. The result is a cleaner reaction profile that simplifies isolation and enhances the overall mass balance of the process.
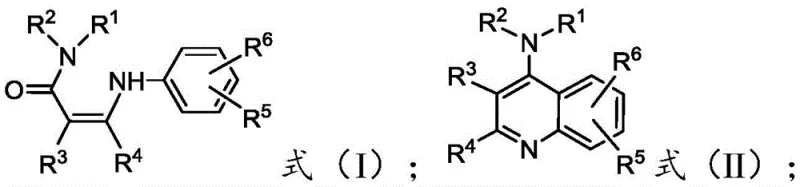
From an impurity control perspective, the absence of metal catalysts is a decisive advantage for regulatory compliance. In traditional metal-catalyzed routes, extensive scavenging steps using silica-bound thiols or other chelating agents are often required to reduce metal levels below ppm thresholds, adding significant cost and time to the manufacturing cycle. The electrophilic activation method described here inherently bypasses this requirement, as the byproducts generated are primarily organic salts that are easily removed during standard aqueous workup procedures. The patent data indicates that this method supports a wide range of substituents at the R1 through R6 positions, including alkyl, alkoxy, halo, and nitro groups, demonstrating exceptional versatility. This structural diversity is critical for optimizing the pharmacokinetic properties of the final drug candidate, allowing for fine-tuning of solubility and metabolic stability without changing the core synthetic strategy.
How to Synthesize 4-Aminoquinoline Compounds Efficiently
Implementing this synthesis route requires careful attention to reagent stoichiometry and temperature control to maximize yield and minimize side reactions. The patent outlines a generalized procedure where the electrophilic activating reagents are first mixed in an organic solvent before the addition of the substrate. This order of addition is critical to ensure the active species is generated in situ prior to encountering the starting material. The reaction progress is typically monitored to ensure complete conversion within the 0.1 to 5.0-hour window, after which a standard quenching and extraction protocol is employed. The detailed standardized synthesis steps see the guide below for specific molar ratios and workup parameters optimized for different substrate classes.
- Mix trifluoromethanesulfonic anhydride and triarylphosphine oxide in an organic solvent such as dichloromethane at temperatures between -15°C and 40°C.
- Add the beta-arylaminoacrylamide precursor (Formula I) to the reaction mixture and stir for 0.1 to 5.0 hours to facilitate intramolecular cyclization.
- Quench the reaction with saline solution, adjust pH to neutral, extract with organic solvent, and purify the resulting 4-aminoquinoline (Formula II) via silica gel chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this metal-free synthesis route translates directly into tangible operational efficiencies and risk mitigation. The primary driver of cost reduction in pharmaceutical intermediate manufacturing is often the complexity of the purification train; by eliminating the need for heavy metal removal steps, this process drastically reduces the consumption of specialized scavenging resins and the associated waste disposal costs. Furthermore, the mild reaction conditions imply lower energy consumption for heating and cooling, contributing to a smaller carbon footprint and reduced utility expenses. The use of readily available starting materials, such as substituted anilines and acrylamides, ensures a stable supply chain that is not subject to the geopolitical volatility often seen with precious metal catalysts. This reliability is paramount for maintaining continuous production schedules and meeting strict delivery timelines for downstream API manufacturers.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts removes a significant line item from the bill of materials, as palladium, copper, or iron salts can be prohibitively expensive at scale. Moreover, the simplified workup procedure reduces the volume of solvents and auxiliary materials required for purification, leading to substantial cost savings in raw material procurement. The higher selectivity of the electrophilic activation method also improves the overall yield, meaning less starting material is wasted to form byproducts, further enhancing the economic viability of the process. These factors combine to create a leaner manufacturing model that maximizes margin potential for high-volume production runs.
- Enhanced Supply Chain Reliability: Sourcing high-purity metal catalysts often involves long lead times and complex logistics, whereas the reagents used in this method, such as triflic anhydride and triphenylphosphine oxide, are commodity chemicals available from multiple global suppliers. This diversification of the supply base reduces the risk of production stoppages due to single-source failures. Additionally, the stability of the reagents allows for bulk purchasing and storage, enabling better inventory management and hedging against price fluctuations. The robustness of the reaction conditions also means that the process is less sensitive to minor variations in raw material quality, providing a buffer against supply chain inconsistencies.
- Scalability and Environmental Compliance: Scaling a metal-free process is inherently safer and more straightforward, as there is no risk of metal accumulation in reactor vessels or wastewater streams. This simplifies the environmental permitting process and reduces the burden on effluent treatment plants, aligning with increasingly stringent global environmental regulations. The ability to run the reaction at near-ambient temperatures also reduces the engineering constraints on reactor design, allowing for the use of standard glass-lined or stainless steel equipment without specialized coatings. This ease of scale-up accelerates the timeline from clinical trial material to commercial launch, ensuring faster time-to-market for new therapeutic candidates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this electrophilic activation technology. These insights are derived directly from the experimental data and claims within the patent documentation, providing a factual basis for decision-making. Understanding these nuances is essential for evaluating the feasibility of integrating this route into existing manufacturing portfolios. The answers reflect the specific advantages of the metal-free approach over legacy methodologies.
Q: Does this synthesis method require transition metal catalysts?
A: No, the method described in patent CN113135852B utilizes an electrophilic activation strategy involving trifluoromethanesulfonic anhydride and triarylphosphine oxide, completely eliminating the need for expensive and toxic transition metal catalysts.
Q: What are the typical reaction conditions for this cyclization?
A: The reaction proceeds under mild conditions, typically ranging from -15°C to 40°C, with reaction times between 0.1 to 5.0 hours, depending on the specific substrate substituents and solvent choice.
Q: How does this method improve impurity profiles compared to traditional routes?
A: By avoiding metal catalysts, this route eliminates the risk of heavy metal residues in the final API intermediate, significantly simplifying the purification process and ensuring compliance with stringent pharmaceutical purity specifications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 4-Aminoquinoline Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic importance of adopting innovative synthetic technologies to maintain a competitive edge in the global pharmaceutical market. Our team of expert process chemists has extensively evaluated the electrophilic activation route described in CN113135852B and confirmed its potential for robust commercial production. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from lab-scale optimization to industrial manufacturing is seamless and efficient. Our state-of-the-art facilities are equipped with rigorous QC labs capable of verifying stringent purity specifications, guaranteeing that every batch of 4-aminoquinoline intermediate meets the highest international standards for safety and efficacy.
We invite you to collaborate with us to leverage this advanced chemistry for your drug development programs. By partnering with our technical procurement team, you can access a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to contact us today to request specific COA data and route feasibility assessments, allowing you to make informed decisions about integrating this high-efficiency synthesis method into your supply chain. Together, we can accelerate the delivery of life-saving antimalarial and therapeutic agents to patients worldwide while optimizing your manufacturing economics.