Advanced Synthesis of Cefdinir Side Chain Acid: A Strategic Upgrade for Global Pharmaceutical Supply Chains
Advanced Synthesis of Cefdinir Side Chain Acid: A Strategic Upgrade for Global Pharmaceutical Supply Chains
The global demand for third-generation cephalosporins continues to drive the need for robust, scalable, and cost-effective synthetic routes for their critical side chains. Patent CN102180843A introduces a transformative preparation method for 2-(2-amino-4-thiazolyl)-2(Z)-trityloxyiminoacetic acid, a pivotal intermediate in the manufacture of Cefdinir. This technology addresses long-standing inefficiencies in prior art by optimizing the O-alkylation step through steric control and replacing hazardous solvents with industrially friendly alternatives. For R&D directors and procurement strategists, understanding this shift is vital, as it directly impacts the purity profile of the final API and the overall cost structure of the supply chain. The structural integrity of this side chain is paramount, as shown in the comparison of active thioester precursors below.
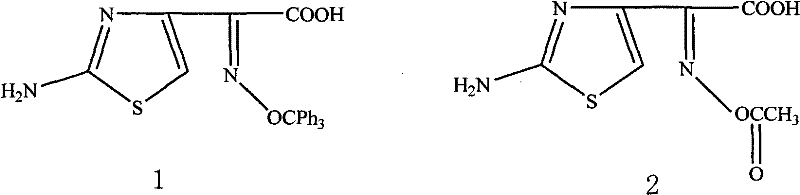
This patent specifically targets the synthesis of the trityl-protected variant, which offers superior stability during the subsequent coupling and deprotection phases compared to acetyl-protected analogs. By securing a reliable cefdinir intermediate supplier who utilizes this advanced methodology, pharmaceutical manufacturers can mitigate risks associated with ring-opening polymerization during the final drug synthesis. The technical breakthrough lies not just in the chemical transformation itself, but in the holistic process design that prioritizes solvent recovery, impurity management, and operational safety, making it a cornerstone for modern beta-lactam production strategies.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of this critical intermediate has been plagued by significant technical and economic hurdles that compromise supply chain efficiency. Early methods, such as those disclosed in WO 03/040116, relied heavily on sulfolane as a solvent, which presents severe downstream processing challenges due to its high boiling point and difficulty in recycling, leading to inflated production costs and environmental burdens. Furthermore, alternative routes described in EP 0355821 utilized sodium hydride, a hazardous and expensive reagent, coupled with dioxane, a known teratogen, creating substantial safety liabilities and regulatory compliance issues for large-scale facilities. Other approaches failed to adequately separate N-alkylated byproducts, resulting in complex mixtures that required extensive and yield-depleting purification steps using toxic solvents like DMF and toluene.
These conventional pathways often suffered from low overall yields, sometimes dropping below 50%, primarily due to the formation of N,O-bis-trityl impurities that were difficult to remove without sacrificing the target product. The reliance on harsh conditions and non-recyclable solvents meant that cost reduction in pharmaceutical intermediates manufacturing was nearly impossible without compromising on quality or safety standards. Consequently, supply chain heads faced unpredictable lead times and inconsistent batch quality, as the purification bottlenecks in these legacy processes frequently caused production delays. The accumulation of these inefficiencies created a pressing need for a method that could deliver high purity without the associated operational hazards and waste generation.
The Novel Approach
The methodology outlined in CN102180843A represents a paradigm shift by introducing a highly selective O-alkylation strategy that fundamentally alters the impurity profile of the reaction. By employing sterically hindered organic strong bases, such as potassium tert-butoxide or sodium tert-butoxide, in solvents like tetrahydrofuran (THF), methyl ethyl ketone (MEK), or acetonitrile, the process effectively suppresses the formation of N-alkylated byproducts. This selectivity is achieved because the bulky tert-butoxy anion favors the formation of the oxime oxygen anion, which then reacts cleanly with chlorotriphenylmethane to form the desired O-trityl ester. The result is a crude product that is significantly cleaner, requiring less aggressive purification and enabling higher overall recovery rates.
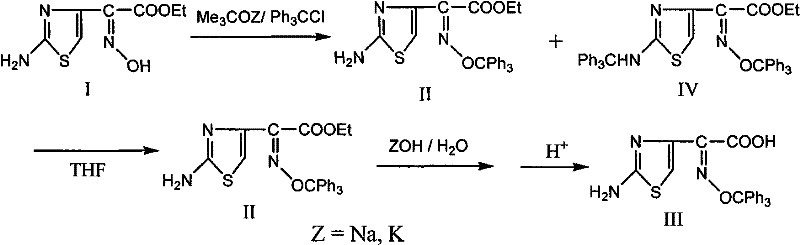
Furthermore, the novel approach incorporates a sophisticated purification step involving recrystallization from THF and water mixtures, which efficiently removes unreacted starting materials and trace impurities without the need for chromatography or toxic solvent exchanges. The subsequent hydrolysis is performed in safe alcohol-water systems, completely eliminating the need for carcinogenic dioxane or high-boiling amides. This streamlined workflow not only enhances the commercial scale-up of complex beta-lactam intermediates but also aligns with green chemistry principles by facilitating solvent recovery. For procurement teams, this translates to a more stable supply of high-purity cefdinir side chain acid with reduced dependency on volatile raw material markets.
Mechanistic Insights into Steric-Controlled O-Alkylation
The core innovation of this patent lies in the precise manipulation of steric hindrance during the alkylation phase to dictate regioselectivity. In traditional nucleophilic substitutions involving ambident nucleophiles like oximes, competition between O-alkylation and N-alkylation is a persistent challenge that often leads to intractable mixtures. However, by utilizing a base with significant steric bulk, such as potassium tert-butoxide, the deprotonation equilibrium is shifted, and the resulting nucleophilic attack is kinetically controlled to favor the oxygen atom. The large tert-butyl group physically obstructs the approach to the nitrogen center or stabilizes the transition state for O-attack, thereby minimizing the generation of the N,O-bis-trityl impurity which is notoriously difficult to separate from the target molecule.
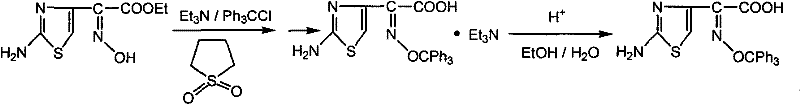
This mechanistic advantage is compounded by the strategic use of aqueous workups immediately following the alkylation reaction. The addition of water or brine allows for the phase separation and removal of unreacted desmethylaminothioxamic acid ethyl ester, which might otherwise carry over into the hydrolysis step and complicate the final isolation. The ability to remove these precursors early in the process flow ensures that the subsequent hydrolysis proceeds with high specificity, yielding the free acid with minimal side reactions. This level of control over the reaction pathway is critical for maintaining the stringent purity specifications required for cephalosporin production, where even trace impurities can affect the crystallization and bioavailability of the final drug product.
Moreover, the crystallization behavior of the intermediate ester is optimized by the choice of solvent systems, leveraging the differential solubility of the O-trityl product versus the N-alkylated impurities. By carefully controlling the temperature and water content during the crystallization from THF or ethyl acetate, the process achieves a purity of greater than 98% before hydrolysis even begins. This pre-hydrolysis purity is a key differentiator, as it reduces the load on the final purification steps and ensures that the hydrolysis byproducts are minimal. For R&D teams, understanding this interplay between base selection, solvent polarity, and crystallization dynamics is essential for replicating the high yields and purity levels demonstrated in the patent examples.
How to Synthesize 2-(2-Amino-4-Thiazolyl)-2(Z)-Trityloxyiminoacetic Acid Efficiently
The synthesis protocol detailed in the patent provides a clear roadmap for transitioning from laboratory scale to commercial production while maintaining rigorous quality control. The process begins with the formation of the oxime anion under inert conditions, followed by the controlled addition of the tritylating agent to manage exotherms and ensure complete conversion. Detailed standardized synthesis steps are provided in the guide below to assist technical teams in implementing this route.
- Perform O-alkylation of ethyl desmethylaminothioxamate using potassium tert-butoxide in THF or MEK to minimize N-alkylation byproducts.
- Purify the crude ester intermediate via recrystallization using tetrahydrofuran and water mixtures to remove unreacted starting materials.
- Execute alkaline hydrolysis in an ethanol-water system followed by acid precipitation to isolate the final high-purity acid product.
Commercial Advantages for Procurement and Supply Chain Teams
Adopting this patented synthesis route offers profound strategic benefits for organizations focused on optimizing their supply chain resilience and cost structures. The elimination of expensive and difficult-to-recycle solvents like sulfolane directly contributes to substantial cost savings in raw material procurement and waste disposal. By switching to common industrial solvents such as THF and methyl ethyl ketone, manufacturers can leverage existing solvent recovery infrastructure, significantly reducing the operational expenditure associated with distillation and solvent make-up. This shift not only lowers the unit cost of the intermediate but also insulates the supply chain from price volatility associated with specialty solvents.
- Cost Reduction in Manufacturing: The replacement of hazardous reagents like sodium hydride with safer, more manageable alkoxides reduces the need for specialized handling equipment and safety protocols, leading to lower overhead costs. Additionally, the high selectivity of the reaction minimizes the loss of valuable starting materials to byproducts, effectively increasing the mass balance efficiency of the entire process. The ability to recycle solvents efficiently further compounds these savings, making the overall cost reduction in pharmaceutical intermediates manufacturing significant and sustainable over the long term.
- Enhanced Supply Chain Reliability: By simplifying the purification process and removing bottlenecks associated with difficult separations, the production cycle time is drastically shortened, allowing for faster turnaround on orders. The use of readily available raw materials and standard solvents ensures that production is not held hostage by the supply constraints of exotic chemicals, thereby enhancing the reliability of the cefdinir intermediate supplier. This robustness is critical for maintaining continuous API production schedules and meeting the demanding delivery timelines of global pharmaceutical clients.
- Scalability and Environmental Compliance: The process is inherently designed for scalability, avoiding exothermic runaways and toxic emissions that often limit batch sizes in traditional methods. The removal of teratogenic solvents like dioxane and high-boiling amides simplifies environmental compliance and reduces the burden on wastewater treatment facilities. This alignment with green chemistry principles not only future-proofs the manufacturing site against tightening regulations but also appeals to increasingly eco-conscious stakeholders in the pharmaceutical value chain.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis method. These insights are derived directly from the comparative data and experimental results presented in the patent documentation, providing clarity on the operational advantages.
Q: How does the new method improve upon the traditional sulfolane-based synthesis?
A: The patented method replaces expensive, high-boiling sulfolane with easily recoverable solvents like THF or methyl ethyl ketone, drastically simplifying solvent recycling and reducing energy consumption during distillation.
Q: What mechanism ensures high purity regarding N-alkylation impurities?
A: By utilizing sterically hindered strong bases such as potassium tert-butoxide, the reaction selectively favors O-alkylation over N-alkylation, significantly suppressing the formation of difficult-to-remove N,O-bis-trityl byproducts.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process avoids hazardous reagents like sodium hydride and teratogenic solvents like dioxane, utilizing standard industrial solvents and mild conditions that are inherently safer and easier to scale for commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-(2-Amino-4-Thiazolyl)-2(Z)-Trityloxyiminoacetic Acid Supplier
At NINGBO INNO PHARMCHEM, we recognize that the quality of your final antibiotic depends heavily on the purity and consistency of your starting materials. As a leading CDMO expert, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated steric-control chemistry described in CN102180843A is executed with precision at every scale. Our rigorous QC labs and stringent purity specifications guarantee that every batch of cefdinir side chain acid meets the exacting standards required for GMP manufacturing, providing you with a secure foundation for your API synthesis.
We invite you to collaborate with our technical procurement team to explore how this advanced route can optimize your specific production needs. By requesting a Customized Cost-Saving Analysis, you can gain a detailed understanding of the potential economic benefits tailored to your volume requirements. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, ensuring that your supply chain is built on the most efficient and reliable chemical technologies available in the market.