Advanced Synthesis of Docarpamine: A Cost-Effective Route for Cardiovascular API Intermediates
Introduction to Advanced Docarpamine Manufacturing
The pharmaceutical industry continuously seeks robust synthetic routes for cardiovascular agents that balance high purity with economic viability. Patent CN1470506A introduces a transformative methodology for the synthesis of Docarpamine, an orally active prodrug of Dopamine Hydrochloride designed to treat conditions such as renal failure, hypertension, and chronic congestive heart failure. Unlike its parent compound, which requires intravenous administration due to poor gastrointestinal absorption and rapid hepatic degradation, Docarpamine offers a stable oral alternative. The core innovation lies in the strategic selection of starting materials and the optimization of reaction conditions to ensure stereochemical fidelity. By utilizing homopiperonylamine as a precursor, the process inherently protects reactive functional groups, thereby streamlining the downstream purification workflow.
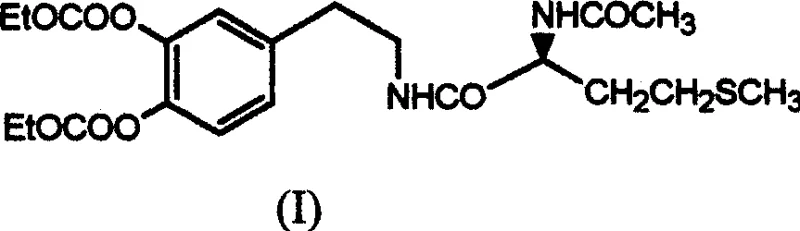
This technical breakthrough addresses the critical challenges associated with catechol chemistry, specifically oxidation and polymerization, which often plague the synthesis of dopamine derivatives. The disclosed method not only enhances the chemical stability of the intermediates but also significantly reduces the operational complexity typically associated with peptide coupling in the presence of sensitive phenolic groups. For R&D directors and process chemists, this represents a shift from laboratory-scale curiosity to a viable commercial manufacturing protocol. The ability to produce high-purity pharmaceutical intermediates without resorting to expensive chromatographic techniques is a substantial value proposition for supply chain stakeholders aiming to reduce lead times and production costs.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies, such as those described in US Patent 4,228,183, relied heavily on the direct use of Dopamine Hydrochloride or its protected derivatives in conjunction with mixed anhydride activation strategies. These conventional routes suffered from significant drawbacks, primarily the tendency for racemization at the chiral center of the amino acid moiety. The formation of mixed acid anhydrides using reagents like vinyl chloroformate often creates highly reactive species that can promote enolization, leading to the loss of optical purity which is critical for biological activity. Furthermore, the direct involvement of free catechol groups in early synthetic stages necessitates rigorous protection and deprotection sequences that add unnecessary steps and reduce overall yield.
Another major bottleneck in traditional synthesis is the reliance on silica gel column chromatography for purification. While acceptable for gram-scale laboratory synthesis, column chromatography is economically and environmentally prohibitive for multi-kilogram or ton-scale production. It consumes vast quantities of organic solvents, generates significant hazardous waste, and limits throughput capacity. The presence of impurities generated from side reactions with unprotected amines or phenols further complicates the isolation process, often resulting in oily residues that are difficult to crystallize. These factors collectively drive up the cost of goods sold (COGS) and extend the manufacturing cycle time, making conventional routes less attractive for cost reduction in pharmaceutical intermediates manufacturing.
The Novel Approach
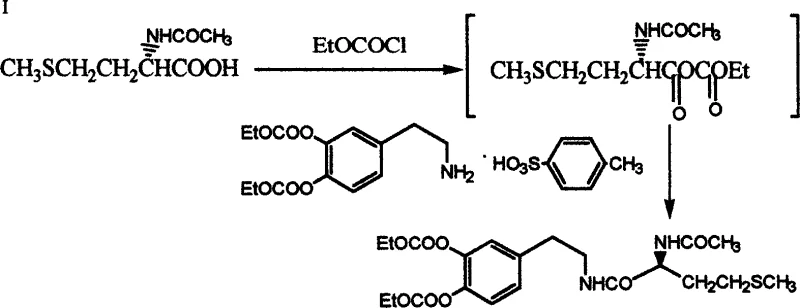
The novel approach detailed in CN1470506A fundamentally reengineers the synthetic logic by reversing the order of functional group manipulation. Instead of starting with dopamine, the process begins with homopiperonylamine, which features a methylenedioxy bridge that effectively masks the two hydroxyl groups intended for the final catechol structure. This structural feature acts as an internal protecting group, rendering the aromatic ring inert to oxidation and electrophilic attack during the critical peptide bond formation. The condensation of N-acetyl-L-methionine with homopiperonylamine proceeds smoothly under mild conditions using coupling agents like DCC and HOBt, yielding a stable crystalline intermediate.
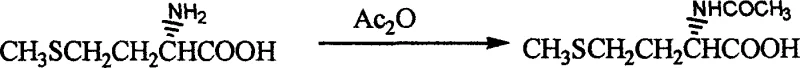
Following the formation of the amide bond, the methylenedioxy group is selectively cleaved using Lewis acids such as boron tribromide (BBr3) or aluminum chloride (AlCl3) to reveal the catechol functionality. This demethylenation step is immediately followed by protection with ethoxycarbonyl groups to finalize the Docarpamine structure. The most significant advantage of this route is the physical state of the intermediates; the key amide intermediate precipitates as a white solid that can be purified by simple recrystallization rather than chromatography. This shift from chromatographic purification to crystallization is a hallmark of a mature, scalable process, enabling commercial scale-up of complex pharmaceutical intermediates with consistent quality and reduced environmental footprint.
Mechanistic Insights into Peptide Coupling and Demethylenation
The success of this synthesis hinges on the precise control of the peptide coupling mechanism. The reaction utilizes a carbodiimide-based activation system, typically Dicyclohexylcarbodiimide (DCC) in the presence of 1-Hydroxybenzotriazole (HOBt). Mechanistically, DCC activates the carboxylic acid of N-acetyl-L-methionine to form an O-acylisourea intermediate. However, O-acylisoureas are prone to rearrangement into unreactive N-acylureas and can cause racemization via oxazolone formation. The addition of HOBt serves a dual purpose: it rapidly converts the O-acylisourea into a more stable and less racemization-prone active ester (the HOBt ester), and it acts as a nucleophilic catalyst. This ensures that the nucleophilic attack by the amine group of homopiperonylamine occurs efficiently at low temperatures, typically between -15°C and 15°C, preserving the chiral integrity of the L-methionine alpha-carbon.
Impurity control is intrinsically built into the molecular design of the starting material. In conventional routes using dopamine, the electron-rich catechol ring is susceptible to oxidative coupling and Michael-type additions, generating polymeric tars that are difficult to remove. By employing homopiperonylamine, the electron density of the aromatic ring is modulated by the methylenedioxy group, and the positions ortho to the nitrogen are blocked. This steric and electronic shielding minimizes side reactions during the exothermic coupling phase. Consequently, the crude reaction mixture contains fewer byproducts, allowing the desired intermediate to crystallize out with high purity upon workup. This mechanistic elegance translates directly to process robustness, as the sensitivity to oxygen and moisture is significantly reduced compared to free catechol handling.
The subsequent demethylenation step employs strong Lewis acids like Boron Tribromide (BBr3) in dichloromethane. The mechanism involves the coordination of the boron atom to the ether oxygens of the methylenedioxy ring, followed by nucleophilic attack by bromide ions to cleave the carbon-oxygen bonds. This reaction must be carefully controlled, typically at 0°C, to prevent over-reaction or degradation of the sensitive amide linkage. Once the free catechol is generated, it is immediately reacted with ethyl chloroformate (Vinyl chloroformate) in the presence of a base like triethylamine. This acylation protects the phenolic hydroxyls as carbonate esters, stabilizing the molecule against oxidation and completing the synthesis of the prodrug. The entire sequence demonstrates a sophisticated understanding of orthogonal protection strategies tailored for industrial application.
How to Synthesize Docarpamine Efficiently
The synthesis of Docarpamine via this novel route is divided into three distinct operational phases: the preparation of the acetylated amino acid, the condensation to form the protected amide intermediate, and the final deprotection-functionalization sequence. Each step has been optimized to maximize yield and minimize impurity carryover. The process begins with the acetylation of L-methionine, followed by the critical coupling reaction with homopiperonylamine hydrochloride using DCC and HOBt in a solvent system such as 1,4-dioxane or dichloromethane. The resulting intermediate is isolated via filtration and washing, avoiding the need for column chromatography. The final transformation involves treating the intermediate with BBr3 to remove the methylene bridge, followed by quenching and protection with ethyl chloroformate to yield the final API intermediate.
- Acetylate L-methionine using acetic anhydride under alkaline conditions to form N-acetyl-L-methionine.
- Condense N-acetyl-L-methionine with homopiperonylamine using a mixed condensing agent system (e.g., DCC/HOBt) to form the key intermediate.
- Perform demethylenation using boron tribromide followed by ethoxycarbonyl protection to yield the final Docarpamine product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this novel synthetic route offers compelling economic and logistical benefits. The primary driver for cost optimization is the substitution of expensive Dopamine Hydrochloride with homopiperonylamine, which is a more abundant and cost-effective commodity chemical. Additionally, the elimination of silica gel chromatography removes a major bottleneck in production throughput. Chromatography is not only solvent-intensive but also requires specialized equipment and extended processing time. By replacing this unit operation with crystallization, the manufacturing cycle time is drastically shortened, and solvent recovery becomes more efficient, leading to substantial cost savings in raw material and waste disposal.
- Cost Reduction in Manufacturing: The economic model of this process is strengthened by the high yield and purity achieved without expensive purification media. The use of standard reagents like DCC and HOBt, which are widely available in the fine chemical market, ensures stable pricing and supply. Furthermore, the ability to purify intermediates via crystallization significantly reduces the volume of organic solvents required per kilogram of product. This reduction in solvent usage directly lowers both the procurement costs for chemicals and the environmental compliance costs associated with solvent waste treatment, contributing to a leaner manufacturing budget.
- Enhanced Supply Chain Reliability: Reliance on complex purification techniques often introduces variability in batch release times. By simplifying the workflow to a series of react-and-crystallize steps, the process becomes more predictable and robust. The starting materials, including N-acetyl-L-methionine and homopiperonylamine, are commercially available from multiple global suppliers, mitigating the risk of single-source dependency. This diversification of the supply base ensures continuity of supply even in the face of market fluctuations, making the production of high-purity pharmaceutical intermediates more resilient to external disruptions.
- Scalability and Environmental Compliance: Scalability is a critical factor for meeting the demands of the global cardiovascular drug market. The described method avoids the use of transition metal catalysts that require stringent removal protocols to meet residual metal specifications in APIs. The absence of heavy metals simplifies the validation process and reduces the analytical burden. Moreover, the process operates at near-ambient temperatures for the coupling step, reducing energy consumption for heating or deep cooling. The overall greener profile of the synthesis, characterized by reduced solvent waste and higher atom economy, aligns well with modern environmental, social, and governance (ESG) goals prevalent in the pharmaceutical industry.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis of Docarpamine. These insights are derived directly from the experimental data and comparative analysis provided in the patent literature. Understanding these nuances is essential for technical teams evaluating the feasibility of adopting this route for commercial production. The answers highlight the specific advantages of the homopiperonylamine strategy over traditional dopamine-based syntheses.
Q: Why is homopiperonylamine preferred over dopamine hydrochloride in this synthesis?
A: Homopiperonylamine is significantly cheaper than dopamine hydrochloride and contains a methylenedioxy group that naturally protects the catechol hydroxyls. This protection prevents oxidation and side reactions during the peptide coupling step, eliminating the need for complex purification methods like silica gel chromatography.
Q: How does this method prevent racemization of the chiral center?
A: The process utilizes mild condensation conditions with HOBt/DCC at low temperatures (-15°C to 15°C). Furthermore, the use of the protected amine substrate avoids the harsh conditions associated with mixed anhydride methods on unprotected catechols, ensuring the stereochemical integrity of the L-methionine moiety is maintained throughout the reaction course.
Q: Is silica gel chromatography required for purification in this industrial process?
A: No, one of the primary advantages of this novel route is the elimination of silica gel chromatography. The key intermediate, N-(N-acetyl-L-methionyl)homopiperonylamine, can be purified via simple crystallization, and the final product is also isolated through crystallization, making the process highly suitable for large-scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Docarpamine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient synthetic routes in the competitive landscape of cardiovascular therapeutics. Our team of expert process chemists has extensively analyzed the methodology disclosed in CN1470506A and possesses the technical capability to implement this advanced synthesis at scale. We understand that transitioning from a laboratory protocol to a commercial process requires meticulous attention to detail, particularly regarding impurity profiling and crystal form control. Our facility is equipped with state-of-the-art reactors and purification units capable of handling the specific solvent systems and temperature profiles required for this chemistry, ensuring that every batch meets the highest standards of quality.
We invite pharmaceutical companies and contract research organizations to collaborate with us on the commercial scale-up of complex pharmaceutical intermediates. Our commitment to excellence is backed by our extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. We maintain stringent purity specifications and operate rigorous QC labs to guarantee the consistency of our products. To discuss how we can support your supply chain needs, please contact our technical procurement team to request a Customized Cost-Saving Analysis. We are ready to provide specific COA data and route feasibility assessments to help you optimize your manufacturing strategy for Docarpamine and related cardiovascular agents.