Scalable Synthesis of Bisacetoxy Deazapurine Derivatives via Palladium-Catalyzed C-H Activation
Scalable Synthesis of Bisacetoxy Deazapurine Derivatives via Palladium-Catalyzed C-H Activation
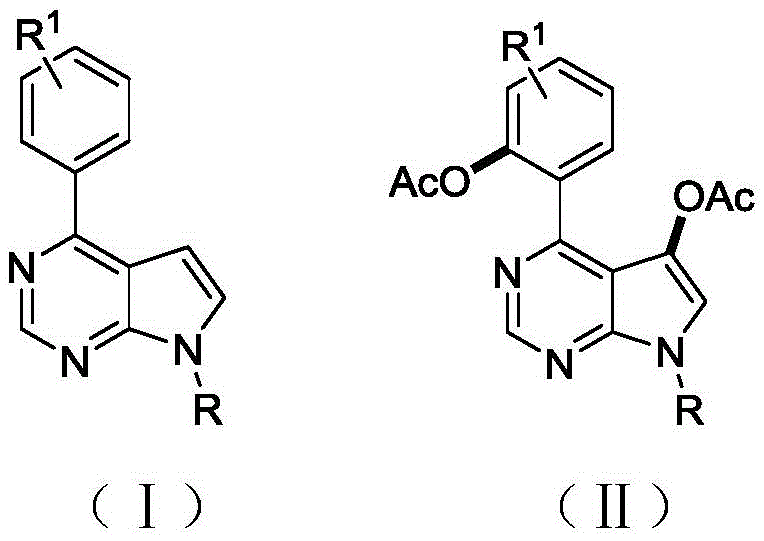
The pharmaceutical industry continuously seeks efficient pathways to access complex heterocyclic scaffolds that serve as critical building blocks for next-generation therapeutics. Patent CN113292563B introduces a groundbreaking methodology for the preparation of bisacetoxy deazapurine derivatives, specifically targeting the pyrrolo[2,3-d]pyrimidine core. This structural motif is renowned for its presence in potent kinase inhibitors and antiviral agents, mimicking natural purine bases while offering enhanced metabolic stability. The disclosed invention establishes a novel synthetic route utilizing palladium-catalyzed direct C-H oxidation to introduce bisacetoxy functional groups at distinct positions simultaneously. This approach represents a significant leap forward in medicinal chemistry, providing a robust platform for the rapid diversification of lead compounds without the need for pre-functionalized starting materials.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the functionalization of pyrrolo[2,3-d]pyrimidine derivatives has been fraught with challenges regarding regioselectivity and reaction severity. Traditional methods often rely on multi-step sequences involving halogenation followed by cross-coupling, which increases waste generation and reduces overall atom economy. Furthermore, existing C-H activation strategies using metals such as iridium, copper, or iron frequently suffer from limited substrate scope, requiring specific directing groups that restrict the chemical diversity of the final products. Many of these prior art methods necessitate harsh reaction conditions, including elevated temperatures exceeding 100°C or the use of toxic oxidants, which pose significant safety risks and complicate downstream purification processes. Additionally, poor regioselectivity often leads to complex mixtures of isomers, drastically lowering yields and increasing the cost of goods sold due to extensive chromatographic separation requirements.
The Novel Approach
In stark contrast, the methodology described in CN113292563B leverages a sophisticated palladium catalytic system to achieve direct double acetoxylation under remarkably mild conditions. By employing iodobenzene diacetate (PhI(OAc)2) as the oxidant and acetic anhydride as the solvent, the reaction proceeds efficiently at temperatures ranging from 50°C to 100°C, with an optimal window around 70°C. This system uniquely enables the simultaneous installation of acetoxy groups at the C-5 position of the pyrrole ring and the ortho-position of the pendant phenyl ring, a transformation that is difficult to achieve with high fidelity using other catalytic systems. The inclusion of sodium iodide as an additive further enhances the catalytic cycle, ensuring high conversion rates and excellent selectivity. This streamlined one-pot procedure eliminates the need for protecting group manipulations and reduces the number of unit operations, thereby significantly enhancing the overall process efficiency for producing high-value pharmaceutical intermediates.
![General reaction scheme showing Pd-catalyzed bisacetoxylation of pyrrolo[2,3-d]pyrimidine derivatives](/insights/img/bisacetoxy-deazapurine-synthesis-pharma-supplier-20260304190644-01.png)
Mechanistic Insights into Pd-Catalyzed C-H Bisacetoxylation
The success of this transformation lies in the intricate interplay between the palladium catalyst and the hypervalent iodine oxidant. The mechanism likely initiates with the coordination of the palladium species to the nitrogen atoms of the pyrrolo[2,3-d]pyrimidine core, facilitating a directed C-H activation at the C-5 position. The presence of the electron-rich pyrrole ring aids in this electrophilic palladation step. Subsequently, the oxidative addition of the hypervalent iodine reagent generates a high-valent Pd(IV) intermediate, which is crucial for the subsequent reductive elimination step that forms the C-O bond. The unique ability of this system to also activate the C-H bond on the adjacent phenyl ring suggests a dual-activation pathway or a sequential oxidation process mediated by the steric and electronic properties of the substrate. The use of acetic anhydride not only serves as the solvent but also as the source of the acetoxy nucleophile, ensuring a high local concentration of the reacting species to drive the equilibrium towards the desired bisacetylated product.
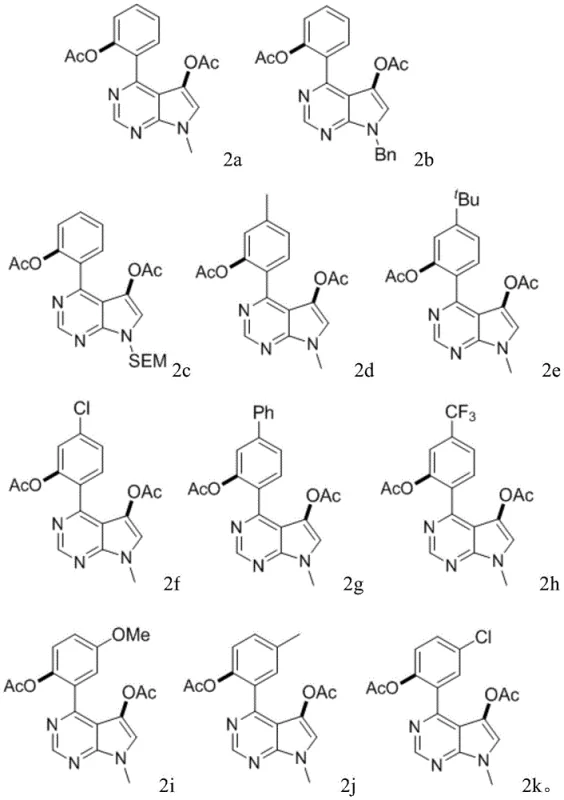
From an impurity control perspective, this catalytic system demonstrates remarkable chemoselectivity, minimizing the formation of mono-acetoxylated byproducts or over-oxidized species. The specific ratio of reagents, particularly the molar ratio of substrate to oxidant (1.0:2.0-4.0) and the catalyst loading (0.01-0.1 equivalents), is finely tuned to suppress side reactions. The reaction tolerates a wide array of functional groups on the phenyl ring, including electron-withdrawing groups like trifluoromethyl and chlorine, as well as electron-donating groups like methyl and methoxy. This tolerance indicates that the catalytic cycle is robust against electronic perturbations, which is vital for maintaining consistent product quality across different batches. The mild acidic environment provided by the acetic anhydride also helps in suppressing base-sensitive degradation pathways, ensuring the integrity of the sensitive heterocyclic core throughout the synthesis.
How to Synthesize Bisacetoxy Deazapurine Derivatives Efficiently
The practical implementation of this synthesis route is designed for operational simplicity, making it highly attractive for process chemistry teams aiming to scale up production. The protocol involves dissolving the starting pyrrolo[2,3-d]pyrimidine compound, iodobenzene diacetate, sodium iodide, and palladium acetate directly into acetic anhydride. The mixture is then heated to the specified temperature range, typically maintained at 70°C for approximately 6 hours to ensure complete conversion. Following the reaction, the workup procedure is straightforward, involving quenching, extraction with ethyl acetate, and standard aqueous washes to remove inorganic salts and residual catalyst. The crude product can be purified using flash column chromatography with a petroleum ether and ethyl acetate gradient, yielding the target bisacetoxy derivatives as stable oily liquids or solids depending on the substituents. Detailed standardized synthesis steps are provided in the guide below.
- Dissolve the pyrrolo[2,3-d]pyrimidine substrate, iodobenzene diacetate, sodium iodide, and palladium acetate in acetic anhydride.
- Stir the reaction mixture at a temperature between 50°C and 100°C, preferably at 70°C, for a duration of 4 to 12 hours.
- Perform post-treatment by extraction with ethyl acetate, washing, drying, and purification via flash column chromatography to isolate the bisacetoxy product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route offers tangible strategic benefits that extend beyond mere technical feasibility. The primary advantage lies in the significant simplification of the supply chain for raw materials; the reagents required, such as palladium acetate and iodobenzene diacetate, are commodity chemicals available from multiple global suppliers, reducing the risk of single-source dependency. Furthermore, the elimination of harsh reaction conditions translates directly into lower energy consumption and reduced wear and tear on reactor vessels, contributing to substantial cost savings in manufacturing overheads. The high selectivity of the reaction minimizes the generation of hazardous waste streams, aligning with increasingly stringent environmental regulations and reducing the costs associated with waste disposal and treatment. This process efficiency allows for faster turnaround times from kilogram-scale development to multi-ton commercial production, ensuring a reliable supply of critical intermediates for downstream drug formulation.
- Cost Reduction in Manufacturing: The streamlined one-pot nature of this reaction eliminates the need for intermediate isolation and purification steps that are typical in traditional multi-step syntheses. By avoiding the use of expensive and specialized ligands often required in other C-H activation protocols, the direct material costs are significantly lowered. The high yield and selectivity reduce the loss of valuable starting materials, improving the overall mass balance and economic viability of the process. Additionally, the use of acetic anhydride as a solvent allows for potential recycling strategies, further driving down the variable costs associated with solvent procurement and disposal in large-scale operations.
- Enhanced Supply Chain Reliability: The broad substrate scope demonstrated in the patent ensures that supply chains remain resilient even when specific starting materials face shortages. Since the method tolerates diverse substituents on the phenyl ring, manufacturers can easily switch between different analogues without re-optimizing the entire process. This flexibility is crucial for maintaining continuous production schedules in the face of fluctuating market demands for specific API intermediates. The robustness of the catalytic system also means that minor variations in raw material quality do not critically impact the reaction outcome, providing a buffer against supply chain volatility and ensuring consistent product availability for pharmaceutical partners.
- Scalability and Environmental Compliance: Scaling this reaction from laboratory to industrial scale is facilitated by the absence of gaseous reagents or extreme pressure requirements, allowing the use of standard stainless steel reactors found in most fine chemical facilities. The mild temperature profile reduces the cooling load and thermal stress on equipment, enhancing operational safety and extending asset life. From an environmental standpoint, the atom economy of introducing acetoxy groups directly via C-H activation is superior to substitution reactions that generate stoichiometric amounts of salt byproducts. This green chemistry attribute supports corporate sustainability goals and simplifies the regulatory approval process for new manufacturing sites by minimizing the environmental footprint of the production facility.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this bisacetoxylation technology. These insights are derived directly from the experimental data and beneficial effects outlined in the patent documentation, providing clarity for R&D and business stakeholders evaluating this technology for integration into their pipelines. Understanding these nuances is essential for making informed decisions about process adoption and long-term sourcing strategies.
Q: What are the key advantages of this Pd-catalyzed bisacetoxylation method?
A: This method operates under mild conditions (50-100°C) using acetic anhydride as both solvent and reagent, offering excellent regioselectivity and broad substrate tolerance compared to traditional harsh oxidation methods.
Q: Which specific compounds showed significant antitumor activity?
A: According to the patent data, compounds 2h (containing a trifluoromethyl group) and 2k (containing a chlorine substituent) demonstrated particularly low IC50 values against JEKO-1 and SU-DHL-4 cell lines, indicating strong potential for anticancer drug development.
Q: Is this synthesis route suitable for large-scale manufacturing?
A: Yes, the process utilizes commercially available reagents like palladium acetate and iodobenzene diacetate, avoids extreme temperatures or pressures, and employs standard workup procedures like extraction and column chromatography, making it highly scalable for industrial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Bisacetoxy Deazapurine Derivative Supplier
As the demand for complex heterocyclic intermediates continues to surge in the oncology and antiviral sectors, partnering with an experienced CDMO is critical for success. NINGBO INNO PHARMCHEM possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from benchtop discovery to full-scale manufacturing. Our state-of-the-art facilities are equipped to handle palladium-catalyzed reactions with stringent purity specifications, supported by rigorous QC labs that utilize advanced analytical techniques to verify identity and potency. We understand the critical nature of supply continuity in the pharmaceutical industry and are committed to delivering high-purity pharmaceutical intermediates that meet the exacting standards of global regulatory bodies.
We invite you to engage with our technical procurement team to discuss how this innovative synthesis route can be tailored to your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain a deeper understanding of the economic benefits of switching to this efficient catalytic method. We encourage potential partners to contact us for specific COA data and route feasibility assessments, allowing us to demonstrate our capability to deliver reliable bisacetoxy deazapurine derivatives that accelerate your drug development timeline.