Scalable Synthesis of Diaziridine-Containing Amino Acids for Advanced Chemical Biology Applications
Scalable Synthesis of Diaziridine-Containing Amino Acids for Advanced Chemical Biology Applications
The rapidly evolving field of chemical biology demands precise molecular tools to map protein-protein interactions and identify drug targets with high fidelity. A significant breakthrough in this domain is detailed in patent CN111087344A, which discloses a novel class of amino acid compounds containing a diaziridine group, specifically designed as potent photoaffinity labeling probes. These molecules, characterized by a unique combination of a photoreactive diaziridine ring and a biologically active amino acid scaffold, offer superior capabilities for affinity-based protein profiling (AfBPP). As a leading entity in the fine chemical sector, we recognize the immense potential of this technology to revolutionize how researchers study intracellular protein dynamics. The patent outlines a robust synthetic methodology that transforms simple, commodity chemicals into high-value research tools, addressing the critical need for reliable pharmaceutical intermediate suppliers who can deliver complex, functionalized building blocks for drug discovery pipelines.
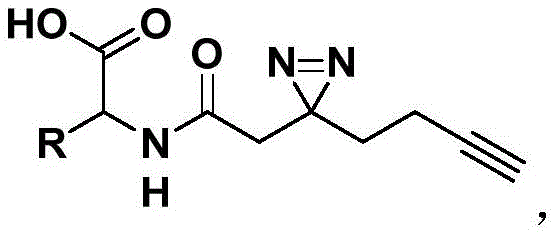
The structural versatility of these compounds allows for the customization of the amino acid moiety (represented by the R group in the general structure), enabling scientists to tailor the probe's binding affinity and specificity for different protein targets. This modularity is essential for developing next-generation therapeutics and diagnostic agents. By integrating the diaziridine photocrosslinker directly onto the amino acid backbone, the resulting conjugates can form irreversible covalent bonds with target proteins upon UV irradiation, stabilizing transient interactions that are otherwise impossible to capture. This capability is paramount for elucidating the mechanisms of action of new drug candidates and understanding complex biological pathways at a molecular level.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this innovation, the synthesis of similar diaziridine-functionalized small molecules often relied on cumbersome and environmentally hazardous protocols. For instance, earlier methodologies described by research groups such as Stephan A. Sieber's utilized 1-hydroxyhept-6-yn-3-one as a starting material, requiring multiple steps to convert ketones into diaziridine structures followed by oxidation. A major drawback of these conventional routes was the reliance on expensive, specialized reagents that were not readily available on the bulk market, driving up the cost of goods significantly. Furthermore, these legacy processes frequently employed heavy metal oxides as oxidants, posing severe environmental risks and necessitating costly waste treatment procedures to remove toxic metal residues from the final product. Such impurities are unacceptable in high-purity API manufacturing or sensitive biological assays, where trace metals can interfere with enzymatic activity or cellular health.
The Novel Approach
In stark contrast, the method disclosed in CN111087344A presents a streamlined, green chemistry approach that drastically simplifies the production workflow. The synthesis begins with ethyl acetoacetate, a ubiquitous and low-cost industrial chemical, which undergoes alkylation with propargyl bromide under the influence of lithium diisopropylamide (LDA) to introduce the necessary alkyne handle. This strategic choice of starting material ensures a stable and economical supply chain foundation. The subsequent steps involve straightforward hydrolysis and a highly efficient diaziridine ring formation using hydroxylamine-O-sulfonic acid and iodine, completely eliminating the need for toxic heavy metal catalysts. The overall route is depicted comprehensively in the reaction scheme below, highlighting the logical progression from simple precursors to the activated NHS ester intermediate.
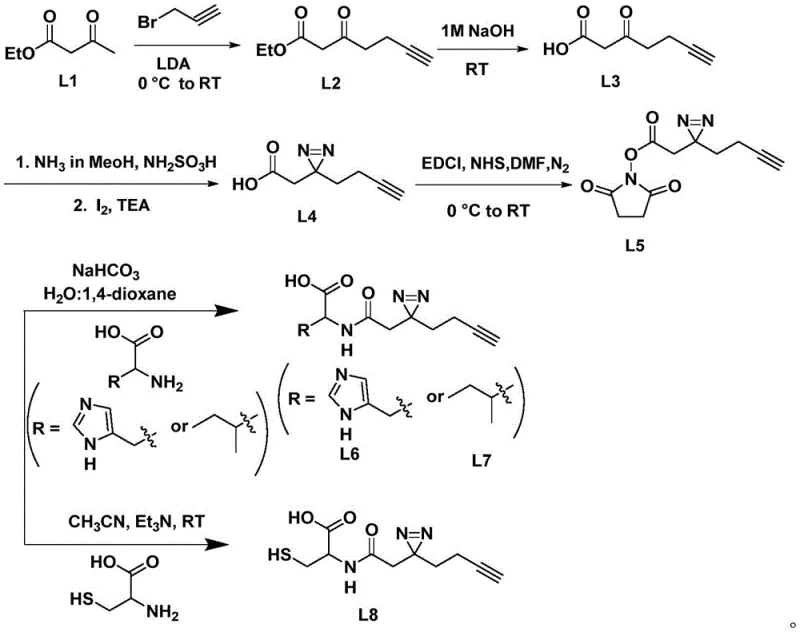
This novel approach not only reduces the number of purification steps—some intermediates like L3 and L4 can be obtained without column chromatography—but also enhances the overall atom economy of the process. By generating an activated N-hydroxysuccinimide (NHS) ester intermediate (L5), the process decouples the complex diaziridine synthesis from the final amino acid coupling. This modular strategy allows manufacturers to produce a single batch of the activated linker and subsequently react it with various amino acids on demand, offering unparalleled flexibility for cost reduction in pharmaceutical intermediate manufacturing. The ability to switch between different amino acid partners without redesigning the entire synthetic pathway is a significant operational advantage for contract development and manufacturing organizations (CDMOs).
Mechanistic Insights into Diaziridine Ring Formation and Amide Coupling
The core chemical transformation in this synthesis is the construction of the diaziridine ring, a three-membered heterocycle containing two nitrogen atoms, which serves as the photoreactive warhead. Mechanistically, this involves the condensation of the ketone intermediate (L3) with hydroxylamine-O-sulfonic acid in an ammonia-methanol solution to form an imine or hydrazone precursor. This is followed by an oxidative cyclization using elemental iodine in the presence of triethylamine. The iodine acts as a mild yet effective oxidant, facilitating the closure of the three-membered ring without degrading the sensitive alkyne functionality present in the side chain. This specific reaction condition is critical; harsher oxidants could potentially react with the triple bond or the carboxylic acid, leading to unwanted byproducts. The precision of this step ensures that the resulting diaziridine (L4) retains high chemical integrity, which is vital for its subsequent photoactivation performance in biological systems.
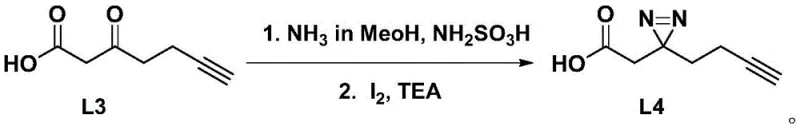
Following the formation of the diaziridine core, the activation of the carboxylic acid group is achieved using EDCI (1-ethyl-(3-dimethylaminopropyl)carbodiimide) and NHS. This converts the relatively unreactive acid into a highly electrophilic NHS ester (L5). The mechanistic advantage of using an NHS ester lies in its stability and reactivity profile; it is stable enough to be isolated and purified, yet reactive enough to couple efficiently with the nucleophilic amine group of amino acids under mild basic conditions. In the final coupling step, the amino acid acts as a nucleophile, attacking the carbonyl carbon of the NHS ester to displace the N-hydroxysuccinimide leaving group, forming a stable amide bond. This bioconjugation strategy is widely regarded as the gold standard in peptide synthesis and ensures that the stereochemistry of the chiral amino acid centers (such as in L-histidine or L-isoleucine) is preserved, preventing racemization which could compromise the biological activity of the final probe.
How to Synthesize Diaziridine-Amino Acid Conjugates Efficiently
The synthesis protocol described in the patent offers a clear, step-by-step guide for producing these high-value intermediates. The process is designed to be operationally simple, utilizing common laboratory solvents like methanol, ethyl acetate, and DMF, which facilitates easy technology transfer from R&D to pilot plant scales. The initial alkylation requires strict temperature control, typically starting at 0°C and warming to room temperature, to manage the exothermic nature of the LDA reaction. Subsequent hydrolysis and diaziridine formation steps are performed under inert atmospheres to prevent moisture interference, ensuring consistent yields. For the final conjugation, the choice of solvent and base is tailored to the specific amino acid; for instance, L-cysteine requires acetonitrile and triethylamine to manage its thiol group, while histidine and isoleucine are coupled in aqueous dioxane with sodium bicarbonate. Detailed standardized synthesis steps for each stage are provided in the guide below.
- Alkylation of ethyl acetoacetate with propargyl bromide using LDA to form the alkyne-keto ester intermediate.
- Hydrolysis of the ester followed by diaziridine ring formation using hydroxylamine-O-sulfonic acid and iodine oxidation.
- Activation of the carboxylic acid as an NHS ester and subsequent coupling with specific amino acids like histidine or cysteine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route represents a strategic opportunity to optimize sourcing costs and mitigate supply risks associated with complex chemical building blocks. The shift away from proprietary, expensive reagents towards commodity chemicals fundamentally alters the cost structure of producing these photocrosslinkers. By leveraging a synthesis path that relies on bulk-available starting materials, manufacturers can achieve substantial cost savings that can be passed down to the end-user, making high-quality proteomics tools more accessible. Furthermore, the robustness of the reaction conditions implies a lower risk of batch failure, which is a critical factor in maintaining supply continuity for long-term research projects.
- Cost Reduction in Manufacturing: The elimination of heavy metal catalysts and expensive specialized reagents significantly lowers the raw material costs. Additionally, the simplified purification process, where intermediates like L3 and L4 do not require column chromatography, reduces solvent consumption and labor hours. This streamlining of the downstream processing directly translates to a lower cost of goods sold (COGS), allowing for more competitive pricing in the global market for research chemicals.
- Enhanced Supply Chain Reliability: Since the synthesis relies on ethyl acetoacetate and propargyl bromide, which are produced on a massive industrial scale, the risk of raw material shortage is minimal. This contrasts sharply with older methods that depended on niche reagents with volatile supply chains. The modular nature of the synthesis, where the activated linker L5 can be stockpiled and reacted with different amino acids as needed, also allows for a more agile response to fluctuating market demands for specific probes.
- Scalability and Environmental Compliance: The process is inherently green, avoiding the generation of heavy metal waste streams that require expensive disposal and regulatory reporting. This environmental compliance simplifies the permitting process for scale-up and aligns with the increasing corporate sustainability goals of major pharmaceutical companies. The reactions are exothermic but manageable, and the use of standard solvents ensures that the process can be safely scaled from gram quantities in the lab to multi-kilogram or tonne scales in commercial reactors without significant re-engineering.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these diaziridine-containing compounds. Understanding these details is crucial for R&D teams evaluating the suitability of these intermediates for their specific protein profiling assays. The answers are derived directly from the technical specifications and experimental data provided in the patent documentation, ensuring accuracy and reliability for decision-making purposes.
Q: What are the primary advantages of this synthesis method over prior art?
A: Unlike previous methods that relied on expensive reagents and heavy metal oxides, this patented route utilizes cheap, commercially available starting materials like ethyl acetoacetate and avoids toxic heavy metals, ensuring a greener and more cost-effective process.
Q: Can this process be scaled for commercial production of proteomics probes?
A: Yes, the synthesis involves robust reactions such as alkylation, hydrolysis, and standard amide coupling which are highly amenable to scale-up. The elimination of complex purification steps for certain intermediates further enhances its feasibility for large-scale manufacturing.
Q: What specific amino acids were successfully conjugated in the patent examples?
A: The patent explicitly demonstrates the successful synthesis of conjugates with L-histidine, L-isoleucine, and L-cysteine, proving the versatility of the activated NHS ester intermediate for coupling with diverse amino acid side chains.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Diaziridine Amino Acid Supplier
As the demand for sophisticated chemical biology tools continues to surge, partnering with a manufacturer that possesses deep technical expertise and scalable infrastructure is essential. NINGBO INNO PHARMCHEM stands at the forefront of this industry, offering extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the sensitive chemistry required for diaziridine synthesis, ensuring that every batch meets stringent purity specifications. We understand that in proteomics research, the quality of the probe dictates the quality of the data; therefore, our rigorous QC labs employ advanced analytical techniques to verify the structural integrity and photo-reactivity of every lot before it leaves our facility.
We invite procurement leaders and R&D directors to collaborate with us to leverage this patented technology for your drug discovery programs. Whether you require custom synthesis of specific diaziridine-amino acid conjugates or bulk supply of the activated NHS ester intermediate, our team is ready to provide a Customized Cost-Saving Analysis tailored to your project volume. Contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us help you accelerate your research with high-performance, reliably sourced chemical intermediates.