Advanced Metal-Free Synthesis of Trifluoroethylselenophenanthridine Derivatives for Commercial Scale-Up
Advanced Metal-Free Synthesis of Trifluoroethylselenophenanthridine Derivatives for Commercial Scale-Up
The pharmaceutical industry is constantly seeking novel scaffolds that can enhance the metabolic stability and bioavailability of lead compounds, and the introduction of fluorine-containing functional groups has become a cornerstone strategy in modern medicinal chemistry. Patent CN110294708B discloses a groundbreaking preparation method for trifluoroethylselenophenanthridine and 3,4-dihydroisoquinoline derivatives, addressing critical challenges in the efficient construction of selenium-containing heterocycles. This technology utilizes a unique combination of isoselenocyanates and trifluoroethyl(aryl)trivalent iodine salts to achieve direct trifluoroethylation and cyclization without the need for transition metal catalysts. For R&D directors and procurement managers alike, this represents a significant leap forward, offering a pathway to high-purity intermediates that are essential for developing next-generation anticancer and antiviral agents. The ability to introduce the -SeCH2CF3 moiety under mild conditions opens new avenues for exploring the biological activity of organoselenium compounds while maintaining strict control over impurity profiles.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of trifluoroethylselenide-containing compounds has been plagued by significant technical hurdles that hindered their widespread adoption in drug discovery pipelines. Previous methodologies, such as those reported by the Feiring group, relied on elimination-addition processes using 2-chloro-2,2-difluoroethyl aryl selenides, which unfortunately generated difluoroolefin by-products that were difficult to separate. Other approaches, like the one developed by Verma's team, utilized diaryl selenides and sodium to generate aryl selenide anions, but these reactions often suffered from low product yields and required harsh reducing conditions. Furthermore, methods employing volatile reagents like CF3CH2I, as seen in work by Weng Zhiqiang's group, posed serious safety and handling challenges due to the low boiling point and high volatility of the trifluoroethylating agent. These limitations not only increased the cost of goods but also introduced variability in batch-to-batch consistency, making scale-up for commercial manufacturing a risky endeavor for supply chain heads.
The Novel Approach
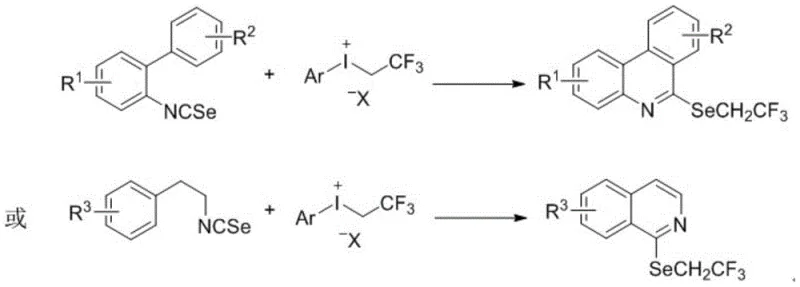
In stark contrast to these legacy methods, the technology described in CN110294708B leverages the high electrophilic activity of trifluoroethyl(aryl)trivalent iodine salts to drive the reaction forward efficiently. By mixing isoselenocyanate with these hypervalent iodine reagents in common organic solvents, the process achieves direct trifluoroethylation and subsequent cyclization in a single pot. This novel approach eliminates the need for transition metal catalysts and additional additives, thereby simplifying the reaction setup and workup procedures significantly. The method operates under mild conditions, with temperatures ranging from -70°C to 100°C and reaction times between 0.5 to 48 hours, allowing for precise control over the reaction kinetics. This robustness ensures that even sensitive functional groups on the aromatic rings remain intact, providing a versatile platform for synthesizing a diverse library of selenium-containing heterocycles with high structural fidelity.
Mechanistic Insights into Hypervalent Iodine-Mediated Cyclization
The core of this synthetic breakthrough lies in the unique reactivity of the trifluoroethyl(aryl)trivalent iodine salt, which acts as a potent electrophilic trifluoroethyl source. The mechanism likely involves the initial attack of the selenium atom in the isoselenocyanate on the electron-deficient carbon of the trifluoroethyl group attached to the iodine center. This interaction facilitates the transfer of the -CH2CF3 group to the selenium, generating a reactive selenonium intermediate. Subsequently, an intramolecular electrophilic aromatic substitution or radical cyclization occurs, closing the ring to form the stable phenanthridine or dihydroisoquinoline skeleton. The absence of transition metals means that the reaction pathway is driven purely by the electronic properties of the hypervalent iodine species, reducing the risk of metal-catalyzed side reactions that often complicate purification. This mechanistic clarity is crucial for R&D teams aiming to optimize reaction parameters for specific substrates, as it allows for rational tuning of electronic effects through substituent modification.
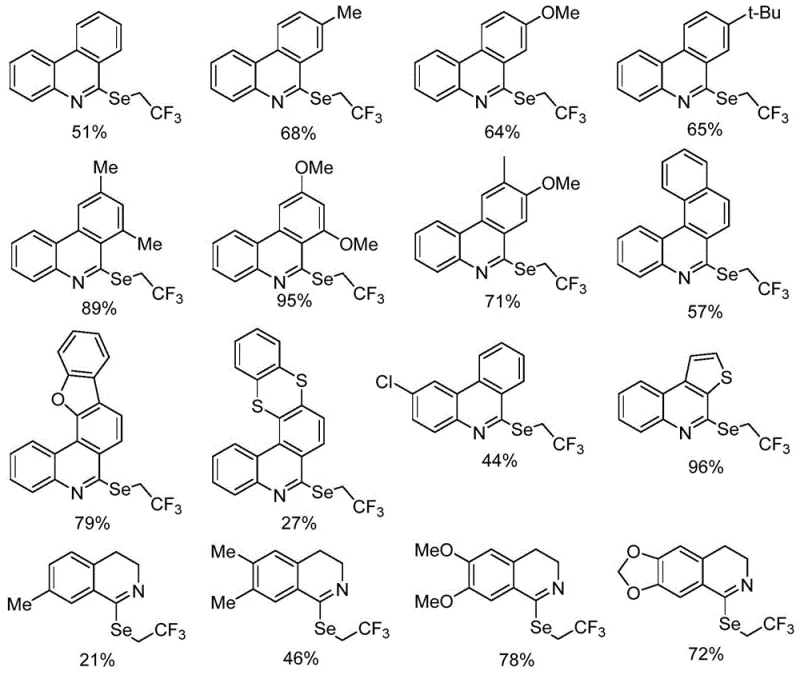
From an impurity control perspective, this metal-free mechanism offers distinct advantages by minimizing the formation of metal-complexed by-products that are notoriously difficult to remove to ppm levels required for API manufacturing. The reaction produces clean profiles, as evidenced by the high yields obtained across a broad range of substrates, including those with electron-donating methoxy groups and electron-withdrawing chloro substituents. The use of standard extraction and silica gel chromatography for purification further underscores the simplicity of the downstream processing. For quality assurance teams, this translates to a more predictable impurity spectrum, facilitating faster method validation and regulatory filing. The ability to tolerate diverse functional groups such as esters, ketones, and halogens without protection-deprotection sequences streamlines the synthetic route, reducing the overall step count and material loss.
How to Synthesize Trifluoroethylselenophenanthridine Efficiently
Implementing this synthesis protocol requires careful attention to the stoichiometry of the hypervalent iodine reagent and the choice of solvent to maximize yield and purity. The patent outlines a straightforward procedure where the isoselenocyanate and the trifluoroethyl(aryl)trivalent iodine salt are mixed in solvents like dichloromethane or acetonitrile under an inert atmosphere. Detailed standardized synthesis steps, including specific molar ratios and workup procedures, are provided in the guide below to ensure reproducibility and safety during laboratory and pilot-scale operations.
- Mix isoselenocyanate substrate with trifluoroethyl(aryl)trivalent iodine salt in an organic solvent such as dichloromethane or acetonitrile.
- Maintain the reaction mixture at a temperature between -70°C and 100°C for a duration ranging from 0.5 to 48 hours depending on substrate reactivity.
- Quench the cooled reaction with saturated NaHCO3, extract with dichloromethane, dry over anhydrous Na2SO4, and purify via silica gel column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this metal-free synthesis technology offers substantial strategic benefits that extend beyond mere chemical efficiency. The elimination of transition metal catalysts directly addresses one of the most costly and time-consuming aspects of pharmaceutical manufacturing: the removal of heavy metal residues. Traditional methods often require specialized scavengers or multiple recrystallization steps to meet stringent ICH Q3D guidelines, adding significant operational expenses and extending lead times. By removing this requirement entirely, the new process drastically simplifies the purification workflow, leading to reduced consumption of auxiliary materials and lower waste disposal costs. This streamlined approach enhances the overall economic viability of producing complex selenium-containing intermediates, making them more accessible for early-stage drug development programs.
- Cost Reduction in Manufacturing: The absence of expensive transition metal catalysts and the associated removal protocols results in a leaner manufacturing process with lower raw material costs. Furthermore, the use of commercially available and stable hypervalent iodine reagents ensures a consistent supply chain without the volatility risks associated with gaseous or highly volatile trifluoroethylating agents. The simplified workup procedure, involving basic extraction and chromatography, reduces the demand for specialized equipment and skilled labor, contributing to significant operational expenditure savings. These factors combined create a more cost-effective production model that can be scaled up without proportional increases in overhead.
- Enhanced Supply Chain Reliability: The robustness of this synthetic method against varying reaction conditions and substrate electronics ensures high batch-to-batch consistency, which is critical for maintaining a reliable supply of key intermediates. The reagents used, such as dichloromethane and common hypervalent iodine salts, are widely available from global chemical suppliers, mitigating the risk of supply disruptions caused by niche reagent shortages. Additionally, the mild reaction conditions reduce the stress on reactor equipment, lowering maintenance requirements and increasing the overall uptime of production facilities. This reliability allows supply chain heads to plan inventory more effectively and respond quickly to fluctuating demand from R&D partners.
- Scalability and Environmental Compliance: Scaling this process from gram to kilogram quantities is facilitated by the lack of exothermic hazards often associated with metal-catalyzed reactions, ensuring safer operation in large-scale reactors. The reduction in hazardous waste generation, particularly heavy metal sludge, aligns with increasingly strict environmental regulations and corporate sustainability goals. The ability to use standard solvents and simple purification techniques means that existing manufacturing infrastructure can be utilized with minimal modification, accelerating the time to market for new drug candidates. This environmental and operational compatibility makes the technology an attractive option for green chemistry initiatives within the pharmaceutical sector.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel synthesis method, drawing directly from the experimental data and beneficial effects outlined in the patent documentation. These insights are designed to clarify the practical implications for process chemists and business stakeholders evaluating this technology for integration into their supply chains.
Q: What are the key advantages of this metal-free synthesis method?
A: The primary advantage is the elimination of transition metal catalysts, which removes the need for expensive and complex heavy metal removal steps, significantly simplifying downstream purification and reducing production costs.
Q: What is the substrate scope for this trifluoroethylation reaction?
A: The method demonstrates excellent functional group compatibility, successfully accommodating substrates with electron-donating groups like methoxy and methyl, as well as electron-withdrawing groups like chloro, with yields ranging from moderate to excellent.
Q: Is this process suitable for large-scale manufacturing?
A: Yes, the use of mild reaction conditions, common organic solvents like dichloromethane, and the absence of sensitive catalysts make this protocol highly scalable for industrial production of pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Trifluoroethylselenophenanthridine Supplier
As the demand for specialized fluorinated and selenated intermediates grows in the oncology and antiviral sectors, having a partner with deep technical expertise is essential for successful project execution. NINGBO INNO PHARMCHEM possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from lab bench to market is seamless and efficient. Our state-of-the-art facilities are equipped with rigorous QC labs capable of meeting stringent purity specifications, guaranteeing that every batch of trifluoroethylselenophenanthridine derivatives meets the highest standards required for clinical and commercial applications. We understand the critical nature of timeline and quality in drug development, and our team is dedicated to providing the support needed to navigate complex regulatory landscapes.
We invite you to engage with our technical procurement team to discuss how this advanced metal-free synthesis can be tailored to your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain a clear understanding of the economic benefits of switching to this efficient protocol for your API manufacturing. We encourage potential partners to contact us for specific COA data and route feasibility assessments, allowing you to make informed decisions based on real-world performance metrics. Let us collaborate to accelerate your drug discovery pipeline with reliable, high-quality chemical solutions.