Advanced Meta-Selective Synthesis of m-Difluoroalkylphenoxypyridine Compounds for Commercial Scale-Up
The pharmaceutical and fine chemical industries are constantly seeking efficient pathways to access structurally complex fluorinated scaffolds, which are critical for enhancing the metabolic stability and bioavailability of modern drug candidates. Patent CN110746349A introduces a groundbreaking preparation method for m-difluoroalkylphenoxypyridine compounds, addressing a long-standing challenge in organic synthesis regarding regioselective C-H bond functionalization. This technology leverages a robust ruthenium-catalyzed system to achieve direct meta-alkylation on the phenoxypyridine core, a transformation that traditionally requires cumbersome multi-step sequences involving blocking groups or specific directing handles. By enabling the direct installation of difluoroalkyl groups at the meta-position, this innovation opens new avenues for the rapid construction of diverse chemical libraries essential for drug discovery programs. The significance of this patent lies not only in its chemical novelty but also in its potential to streamline the supply chain for high-value pharmaceutical intermediates, offering a reliable alternative to conventional synthetic routes that often suffer from poor atom economy and low overall yields.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the functionalization of phenoxypyridine derivatives has been heavily constrained by the inherent electronic properties of the aromatic ring system. Traditional electrophilic aromatic substitution reactions are strongly governed by the directing effects of the existing substituents, typically favoring ortho- and para-positions due to resonance stabilization of the intermediate sigma complexes. Consequently, accessing the meta-position on a phenoxypyridine scaffold using classical chemistry is exceptionally difficult and often impossible without resorting to indirect strategies. These conventional strategies frequently involve the installation of temporary blocking groups, followed by the desired transformation, and finally, a deprotection step to reveal the target meta-substituted product. Such multi-step protocols inevitably lead to increased material consumption, higher waste generation, and significantly reduced overall yields, which negatively impacts the cost-efficiency of the manufacturing process. Furthermore, the use of harsh reagents often required for these indirect routes can compromise the integrity of sensitive functional groups present on the molecule, limiting the scope of substrates that can be successfully processed.
The Novel Approach
In stark contrast to these legacy methods, the technology disclosed in CN110746349A utilizes a transition metal-catalyzed C-H activation strategy that bypasses the electronic limitations of the substrate. By employing ruthenium trichloride in conjunction with a specific carboxylic acid additive, the reaction system is able to override the natural ortho/para directing bias and selectively functionalize the meta-position. This direct approach eliminates the need for pre-functionalization or protecting group chemistry, collapsing what was once a three or four-step sequence into a single, operationally simple transformation. The use of ethyl 2-bromodifluoroacetate as the alkylating agent allows for the efficient introduction of the difluoroethyl ester moiety, a valuable pharmacophore in medicinal chemistry. This novel approach not only simplifies the synthetic workflow but also enhances the sustainability of the process by reducing the number of unit operations and the associated solvent and reagent consumption, aligning perfectly with the principles of green chemistry and modern manufacturing efficiency.
Mechanistic Insights into Ru-Catalyzed Meta-Selective C-H Functionalization
The success of this transformation hinges on the intricate interplay between the ruthenium catalyst and the adamantanecarboxylic acid additive, which together facilitate a unique catalytic cycle. It is hypothesized that the ruthenium species coordinates with the pyridine nitrogen atom, acting as a weak directing group to bring the metal center into proximity with the aromatic ring. However, unlike strong directing groups that force ortho-activation, the specific ligand environment created by the bulky adamantanecarboxylic acid additive sterically hinders the ortho-positions. This steric pressure, combined with the electronic modulation of the metal center, directs the C-H activation event to the more accessible and electronically favorable meta-position. The subsequent reaction with the bromodifluoroacetate likely proceeds through a radical or organometallic intermediate, where the carbon-fluorine bonds are preserved while the carbon-bromine bond is cleaved to form the new carbon-carbon bond. This mechanistic pathway is highly sensitive to reaction conditions, requiring precise control over temperature and stoichiometry to maintain catalyst turnover and prevent side reactions such as homocoupling or over-alkylation.
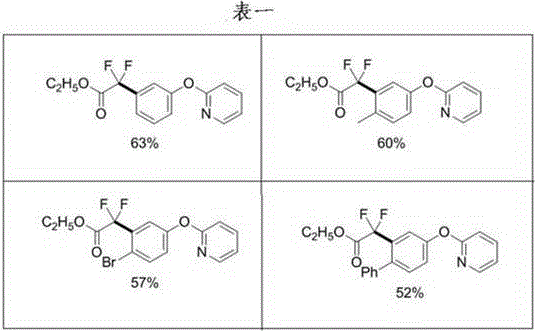
Controlling the impurity profile in such complex C-H functionalization reactions is paramount for pharmaceutical applications, where strict regulatory standards dictate the quality of intermediates. The patent data indicates that the reaction conditions, specifically the temperature range of 105-120°C and the reaction time of 20-28 hours, are optimized to maximize the conversion of the starting material while minimizing the formation of regioisomeric byproducts. The use of potassium carbonate as a base serves to neutralize the hydrobromic acid generated during the reaction, preventing acid-catalyzed decomposition of the sensitive difluoroester group. Furthermore, the choice of solvent, such as 1,2-dichloroethane or 1,4-dioxane, plays a critical role in solubilizing the organic substrates and stabilizing the catalytic species throughout the extended reaction period. The resulting crude mixture, as evidenced by the yields ranging from 52% to 63% across different substrates, suggests a clean reaction profile that facilitates downstream purification via standard column chromatography, ensuring the final product meets the high-purity specifications required for downstream drug synthesis.
How to Synthesize m-Difluoroalkylphenoxypyridine Efficiently
The implementation of this synthesis route in a laboratory or pilot plant setting requires strict adherence to the optimized parameters defined in the patent to ensure reproducibility and safety. The process begins with the precise weighing of the phenoxypyridine substrate and the difluoroalkylating reagent, maintaining a molar ratio that favors the complete consumption of the valuable substrate. The addition of the ruthenium catalyst and the adamantanecarboxylic acid additive must be performed under an inert atmosphere to prevent oxidation of the metal center, which could lead to catalyst deactivation. Once the reagents are charged into the pressure reaction tube along with the solvent and base, the system is sealed and heated to the target temperature, where it is maintained with vigorous stirring to ensure homogeneous heat transfer and mass transfer throughout the reaction mixture. Detailed standardized synthesis steps see the guide below.
- Charge the reactor with phenoxypyridine substrate, ethyl 2-bromodifluoroacetate, RuCl3 catalyst, K2CO3 base, and adamantanecarboxylic acid additive in 1,2-dichloroethane.
- Seal the reaction vessel under nitrogen atmosphere and heat the mixture to a controlled temperature range of 105-120°C with continuous stirring.
- Maintain reaction conditions for 20-28 hours to ensure complete conversion, followed by standard column chromatography separation to isolate the target meta-substituted product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthetic route offers substantial strategic benefits that extend beyond simple chemical yield. The primary advantage lies in the drastic simplification of the manufacturing process, which directly translates to reduced operational complexity and lower capital expenditure requirements. By eliminating multiple synthetic steps, the facility can reduce the number of reactors needed for a specific campaign, thereby increasing overall plant throughput and flexibility. This efficiency gain is critical in a competitive market where speed to market and cost competitiveness are key differentiators. Furthermore, the use of readily available starting materials such as phenoxypyridines and ethyl 2-bromodifluoroacetate ensures a stable supply chain, reducing the risk of bottlenecks associated with exotic or custom-synthesized reagents. The robustness of the reaction conditions also implies a wider operating window, making the process more forgiving and easier to control at a commercial scale, which minimizes the risk of batch failures and production delays.
- Cost Reduction in Manufacturing: The elimination of protection and deprotection steps significantly reduces the consumption of auxiliary reagents and solvents, leading to a lower cost of goods sold. By avoiding the use of expensive palladium catalysts often required for similar transformations and utilizing more abundant ruthenium sources, the raw material costs are optimized. Additionally, the one-pot nature of the reaction reduces labor costs and energy consumption associated with heating, cooling, and transferring materials between multiple unit operations. These cumulative savings allow for a more competitive pricing structure for the final intermediate, providing a distinct margin advantage for downstream drug manufacturers.
- Enhanced Supply Chain Reliability: The reliance on commodity chemicals and standard reaction conditions enhances the resilience of the supply chain against external disruptions. Since the reagents are not proprietary or difficult to source, procurement teams can easily qualify multiple vendors for raw materials, mitigating the risk of single-source dependency. The simplified workflow also shortens the overall production lead time, allowing for faster response to fluctuating market demands and urgent orders from pharmaceutical clients. This agility is crucial for maintaining continuous supply in the fast-paced pharmaceutical industry, where delays can have significant downstream impacts on clinical trial timelines.
- Scalability and Environmental Compliance: The process is inherently scalable due to its simple operational requirements and the absence of hazardous intermediates that often complicate scale-up. The reduced number of steps inherently lowers the total waste generation, aligning with increasingly stringent environmental regulations and corporate sustainability goals. The use of standard solvents that can be recovered and recycled further minimizes the environmental footprint of the manufacturing process. This compliance with green chemistry principles not only reduces waste disposal costs but also enhances the corporate image of the manufacturer as a responsible partner in the global pharmaceutical supply chain.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation and benefits of this meta-selective difluoroalkylation technology. These answers are derived directly from the experimental data and technical specifications provided in the patent documentation, ensuring accuracy and relevance for technical decision-makers. Understanding these details is essential for evaluating the feasibility of integrating this route into existing production portfolios or R&D pipelines. The information provided here serves as a foundational guide for further technical discussions and feasibility assessments.
Q: What is the primary advantage of this Ru-catalyzed method over traditional electrophilic substitution?
A: Traditional electrophilic substitution on phenoxypyridine scaffolds predominantly yields ortho- or para-substituted products due to electronic directing effects. This novel ruthenium-catalyzed protocol uniquely achieves meta-selectivity, accessing chemical space that is otherwise difficult to reach without multi-step protection-deprotection sequences.
Q: What are the critical reaction conditions for optimal yield in this difluoroalkylation process?
A: The process requires a specific combination of ruthenium trichloride as the catalyst and adamantanecarboxylic acid as a crucial additive. The reaction must be conducted in solvents like 1,2-dichloroethane or 1,4-dioxane at elevated temperatures between 105°C and 120°C for a duration of 20 to 28 hours to drive the C-H functionalization to completion.
Q: Is this synthesis route suitable for large-scale commercial manufacturing?
A: Yes, the methodology is designed as a one-step direct functionalization, which significantly simplifies the operational workflow compared to traditional multi-step routes. The use of commercially available reagents and standard heating conditions facilitates scalability, making it a viable candidate for industrial production of complex pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable m-Difluoroalkylphenoxypyridine Supplier
The technological potential of the Ru-catalyzed meta-selective functionalization described in CN110746349A represents a significant leap forward in the synthesis of complex fluorinated intermediates. At NINGBO INNO PHARMCHEM, we possess the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that this innovative chemistry can be seamlessly transferred from the laboratory to the manufacturing plant. Our state-of-the-art facilities are equipped with rigorous QC labs and stringent purity specifications to guarantee that every batch of m-difluoroalkylphenoxypyridine meets the highest industry standards. We understand the critical nature of these intermediates in the drug development lifecycle and are committed to delivering consistent quality and reliability to our global partners.
We invite procurement leaders and R&D directors to engage with our technical procurement team to discuss how this specific synthesis route can optimize your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of switching to this streamlined process. We encourage you to contact us to obtain specific COA data and route feasibility assessments tailored to your project requirements. Our team is ready to support your transition to more efficient and cost-effective manufacturing solutions, ensuring your projects stay on schedule and within budget.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →