Advanced Manufacturing of Diazaspiro Compounds for Pharmaceutical Applications
The pharmaceutical industry continuously seeks robust synthetic pathways for complex heterocyclic scaffolds, particularly diazaspiro compounds which serve as critical building blocks for next-generation therapeutics. Patent CN102267995A introduces a groundbreaking methodology for preparing these structurally rigid molecules, specifically targeting the synthesis of compounds represented by Formula I and Formula VI. These spirocyclic architectures are increasingly recognized for their ability to improve the metabolic stability and binding affinity of drug candidates, ranging from fluoroquinolone antibiotics to peptidomimetics used in treating metabolic disorders. The innovation lies in a streamlined approach that bypasses the cumbersome multi-step sequences typical of legacy chemistry, offering a direct route from readily available cyclic amino acid derivatives. By leveraging a strategic combination of deprotonation, alkylation, and azide-mediated cyclization, this technology addresses the longstanding challenges of low throughput and high operational costs associated with spirocycle manufacturing.
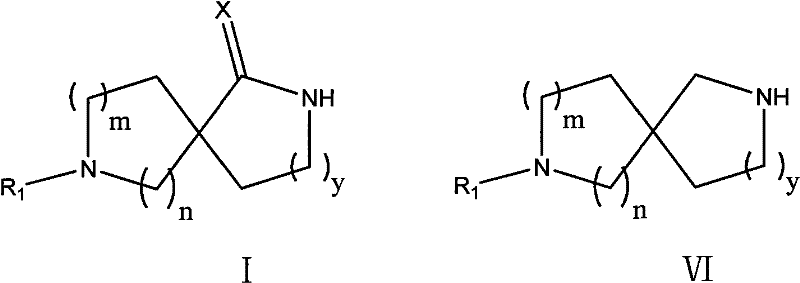
The significance of this technological advancement cannot be overstated for R&D teams focused on rapid lead optimization. Traditional methods often struggle with the steric congestion inherent in forming quaternary spiro-centers, leading to poor conversion rates and difficult purification profiles. The disclosed method overcomes these kinetic barriers by utilizing highly reactive lithiated intermediates that facilitate efficient ring closure. Furthermore, the versatility of the route allows for the modulation of ring sizes (where m and n vary from 1 to 3), providing a versatile platform for generating diverse libraries of spirocyclic amines and lactams. This flexibility is crucial for medicinal chemists aiming to explore structure-activity relationships (SAR) around the spiro-junction without being constrained by synthetic feasibility. As we delve deeper into the mechanistic nuances and commercial implications, it becomes clear that this patent represents a pivotal shift towards more sustainable and economically viable production of high-value pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this invention, the synthesis of diazaspiro compounds was plagued by inefficiencies that severely hampered commercial scalability. As documented in the background art, existing methodologies such as those described in J. Med. Chem. (1990) suffered from abysmal overall yields, sometimes as low as 2.6%, rendering them practically useless for large-scale production. These conventional routes typically relied on harsh reaction conditions, including cryogenic temperatures and the use of expensive, moisture-sensitive reagents that demanded rigorous anhydrous environments. A major bottleneck was the necessity for multiple chromatographic purification steps between each transformation, which not only consumed vast quantities of silica gel and solvents but also resulted in significant material loss at every stage. For instance, earlier schemes often required the isolation of unstable intermediates, increasing the risk of decomposition and complicating the supply chain logistics for active pharmaceutical ingredient (API) manufacturers.
Furthermore, the reliance on specific protecting group strategies in older methods added unnecessary synthetic bulk, requiring additional steps for installation and removal that contributed nothing to the final molecular complexity. The cumulative effect of these inefficiencies was a process with a large environmental footprint and prohibitive costs, making it difficult for procurement managers to justify the sourcing of such intermediates for clinical trials, let alone commercial launch. The inability to telescope reactions meant that production timelines were extended, creating vulnerabilities in the supply chain where delays in one purification step could halt the entire manufacturing campaign. Consequently, there was a pressing industry need for a method that could simplify the topology of the synthesis while simultaneously improving the mass balance and operational safety profile.
The Novel Approach
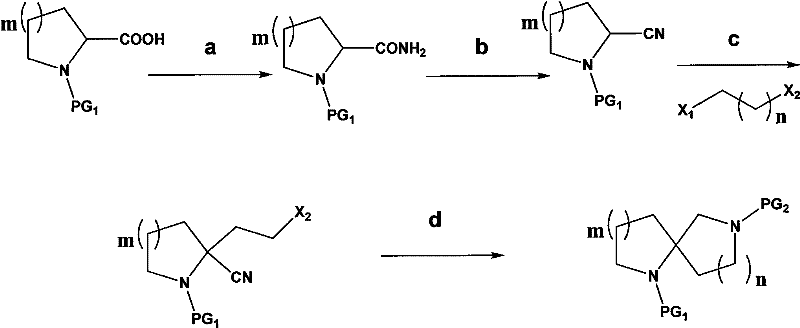
The methodology outlined in CN102267995A presents a paradigm shift by condensing the synthesis into a concise sequence that maximizes atom economy and operational simplicity. The core innovation involves the use of a dihaloalkane to bridge the nitrogen atom of a cyclic amino acid ester, effectively constructing the second ring in a single alkylation event followed by cyclization. Unlike the fragmented approaches of the past, this novel route allows for the direct conversion of the alkylated intermediate into the azide species without isolation, a technique known as telescoping that drastically reduces solvent usage and processing time. The subsequent reduction-cyclization step utilizes triphenylphosphine and water, mild reagents that safely convert the azide functionality into the requisite amine which then spontaneously closes the ring to form the stable spiro-lactam structure. This approach eliminates the need for column chromatography for the majority of intermediates, relying instead on crystallization or simple extraction workups.
From a process chemistry perspective, the new method offers remarkable robustness, operating effectively within a temperature range of -30°C to 85°C, which is far more manageable than the deep cryogenic conditions required by competing technologies. The overall yield for the formation of the spirocyclic lactam (Formula I) has been improved to a range of 40% to 55%, representing a substantial increase over the historical benchmarks of 2.6% to 36%. This improvement is not merely incremental; it fundamentally alters the cost structure of the molecule, making it accessible for broader therapeutic applications. Additionally, the final reduction step to obtain the diamine (Formula VI) proceeds with high efficiency (70% to 90% yield) using borane or lithium aluminum hydride, ensuring that both the lactam and diamine variants of the scaffold are readily available. This dual-output capability enhances the value proposition for pharmaceutical partners who may require either structural motif for different stages of drug development.
Mechanistic Insights into LDA-Mediated Alkylation and Azide Cyclization
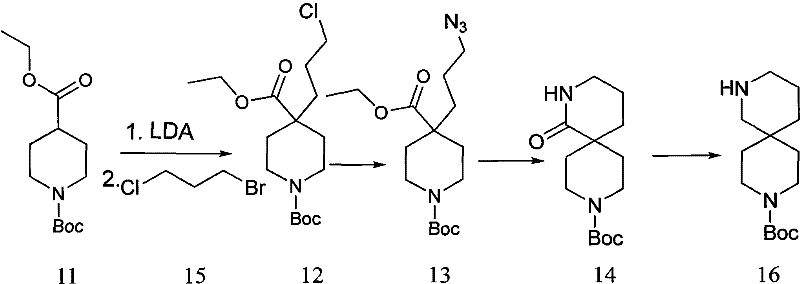
The success of this synthetic route hinges on the precise control of nucleophilicity and regioselectivity during the initial alkylation phase. The process initiates with the deprotonation of the alpha-carbon adjacent to the ester or amide carbonyl in the starting material (Formula V) using lithium diisopropylamide (LDA). This strong, non-nucleophilic base generates a resonance-stabilized enolate that acts as a potent nucleophile. When reacted with a terminal dihaloalkane (Formula III), the enolate selectively displaces the more reactive halogen atom, forming the carbon-carbon bond that extends the side chain. The choice of dihaloalkane is critical; the patent specifies using halogens with differentiated reactivity (e.g., bromine and chlorine) to ensure mono-alkylation occurs at the desired position without polymerizing or forming bis-alkylated byproducts. This regiocontrol is essential for setting up the correct chain length required for the subsequent ring closure, preventing the formation of incorrect ring sizes that would act as difficult-to-remove impurities.
Following alkylation, the introduction of the second nitrogen atom is achieved through a classic SN2 substitution using sodium azide. The remaining halogen on the side chain serves as a leaving group, allowing the azide anion to displace it and install the latent amine functionality. The true elegance of the mechanism is revealed in the final cyclization step. Upon treatment with triphenylphosphine, the azide group undergoes a Staudinger reduction to form an iminophosphorane intermediate, which is subsequently hydrolyzed to the primary amine. In the presence of base (such as potassium carbonate) and heat, this newly formed amine attacks the electrophilic carbonyl carbon of the original ester moiety. This intramolecular nucleophilic acyl substitution results in the expulsion of the alkoxide leaving group and the formation of the thermodynamically stable five- or six-membered lactam ring fused at the spiro-center. This cascade effectively locks the conformational freedom of the molecule, creating the rigid diazaspiro backbone that is highly prized in drug design for its ability to project pharmacophores in specific spatial orientations.
How to Synthesize Diazaspiro Compounds Efficiently
The practical implementation of this synthesis requires careful attention to stoichiometry and temperature control to maximize the benefits of the telescoped process. The procedure begins with the generation of the lithiated species under inert atmosphere, followed by the controlled addition of the dihaloalkane to manage exotherms. Once the alkylation is complete, the reaction mixture is not worked up but is instead concentrated and redissolved for the direct addition of sodium azide, capitalizing on the crude intermediate's reactivity. The final cyclization is driven by thermal energy and base catalysis, pushing the equilibrium towards the closed ring system. For a comprehensive understanding of the specific molar ratios, solvent choices, and workup procedures detailed in the patent examples, please refer to the standardized guide below.
- Deprotonate the starting cyclic amino acid ester using LDA at low temperature, followed by alkylation with a dihaloalkane to form the first intermediate.
- Perform nucleophilic substitution on the halogenated intermediate using sodium azide to introduce the nitrogen source for the second ring.
- Execute a reduction cyclization using triphenylphosphine and water, followed by base-mediated ring closure to yield the final diazaspiro lactam.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route translates into tangible strategic advantages that extend beyond simple unit cost savings. The elimination of column chromatography is perhaps the most significant factor, as this unit operation is notoriously resource-intensive, requiring large volumes of high-purity solvents and specialized silica media that drive up both direct material costs and waste disposal fees. By replacing chromatography with crystallization and extraction, the process becomes inherently more scalable, allowing for the use of standard stainless steel reactors rather than specialized glass-lined vessels often needed for corrosive or sensitive chromatographic fractions. This simplification reduces the capital expenditure required for manufacturing suites and lowers the barrier to entry for contract manufacturing organizations (CMOs) looking to bid on production contracts for these intermediates.
- Cost Reduction in Manufacturing: The streamlined nature of the reaction sequence directly impacts the cost of goods sold (COGS) by minimizing the number of discrete unit operations. With fewer isolation steps, there is a corresponding reduction in labor hours, equipment occupancy time, and utility consumption (heating/cooling). The use of commodity chemicals like LDA, sodium azide, and triphenylphosphine ensures that raw material costs remain stable and predictable, shielding the supply chain from the volatility often associated with exotic transition metal catalysts. Furthermore, the higher overall yield means that less starting material is required to produce the same amount of final product, effectively amplifying the purchasing power of the raw material budget and reducing the cost per kilogram of the active intermediate significantly.
- Enhanced Supply Chain Reliability: Supply continuity is bolstered by the robustness of the chemistry, which tolerates a wider range of operating conditions compared to fragile cryogenic processes. The ability to telescope steps reduces the inventory of hazardous intermediates that need to be stored or transported between facilities, thereby lowering regulatory compliance burdens and insurance costs. Since the reagents are widely available from multiple global suppliers, the risk of single-source bottlenecks is mitigated. This resilience is critical for maintaining uninterrupted production schedules for downstream API manufacturing, ensuring that clinical trial materials and commercial stockpiles can be replenished rapidly in response to market demand without the long lead times associated with complex multi-step syntheses.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, the process offers a greener profile by reducing the E-factor (mass of waste per mass of product). The avoidance of heavy metal catalysts eliminates the need for expensive and technically challenging metal scavenging steps to meet strict residual metal specifications in pharmaceutical products. The solvent systems employed, primarily THF, DMF, and ethyl acetate, are well-understood and can be efficiently recovered and recycled using standard distillation infrastructure. This alignment with green chemistry principles not only supports corporate sustainability goals but also simplifies the regulatory filing process, as the impurity profile is cleaner and easier to characterize, facilitating faster approval from health authorities.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this diazaspiro synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, aimed at clarifying the operational benefits for potential licensees and manufacturing partners. Understanding these nuances is key to evaluating the fit of this technology within your existing production portfolio.
Q: What are the primary yield improvements in this new diazaspiro synthesis method?
A: The patented method achieves an overall yield of 40% to 55% for the spirocyclic lactam, significantly outperforming prior art methods which ranged from 2.6% to 36%.
Q: Does this process require expensive transition metal catalysts?
A: No, the process utilizes common organic reagents such as LDA, sodium azide, and triphenylphosphine, avoiding the need for costly precious metal catalysts often found in alternative routes.
Q: Is intermediate purification necessary between reaction steps?
A: The method allows for telescoping, where the crude product from the alkylation step can be directly used in the azide substitution without isolation, reducing solvent waste and processing time.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Diazaspiro Compound Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality spirocyclic intermediates play in the development of life-saving medications. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and efficient. We are committed to delivering products with stringent purity specifications, utilizing our rigorous QC labs to verify that every batch meets the exacting standards required by global regulatory agencies. Our facility is equipped to handle the specific reagents and conditions outlined in this patent, guaranteeing a consistent supply of diazaspiro compounds that empower your R&D efforts.
We invite you to collaborate with us to leverage this advanced synthetic technology for your next project. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can optimize your supply chain and accelerate your time to market. Let us be your partner in turning complex chemical challenges into commercial successes.