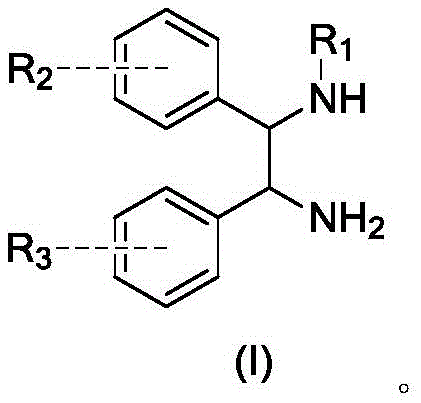
Advanced Cyclization Strategy for High-Purity Mono-Substituted Amines in Commercial Scale-Up
Advanced Cyclization Strategy for High-Purity Mono-Substituted Amines in Commercial Scale-Up
The pharmaceutical industry constantly seeks robust synthetic routes for chiral intermediates, particularly those derived from symmetric diamines like 1,2-diphenylethylenediamine. Patent CN110066233B introduces a transformative preparation method for mono-substituted amine compounds that addresses the longstanding challenge of selectivity in symmetric systems. This technology leverages a strategic cyclization-protection sequence followed by selective ring-opening and deprotection to achieve yields and purities that conventional direct substitution methods cannot match. By converting the reactive diamine into a cyclic imidazoline intermediate, the process effectively differentiates the two amino groups, allowing for precise mono-functionalization. This breakthrough is critical for manufacturers aiming to produce high-value chiral catalysts and active pharmaceutical ingredients where impurity profiles are strictly regulated. The method demonstrates exceptional versatility, accommodating various leaving groups such as trifluoromethylsulfonyl and p-toluenesulfonyl, while maintaining the stereochemical integrity of the chiral centers throughout the transformation.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthetic strategies for functionalizing symmetric diamines often suffer from poor regioselectivity, leading to complex mixture of products that are costly and difficult to separate. When a leaving group is introduced directly to a diamine substrate without protection, the statistical probability favors the formation of disubstituted species alongside the desired mono-substituted target. As illustrated in the comparative data, direct reaction conditions frequently result in disubstituted impurity levels exceeding 30%, rendering the crude material unsuitable for downstream pharmaceutical applications without extensive purification. This lack of selectivity not only depresses the overall yield of the target molecule but also necessitates resource-intensive chromatographic separations or multiple recrystallization steps to meet purity specifications. Furthermore, the presence of structurally similar disubstituted impurities can complicate the crystallization behavior of the product, leading to oiling out or the formation of solvates that trap impurities, thereby compromising the quality of the final active ingredient.
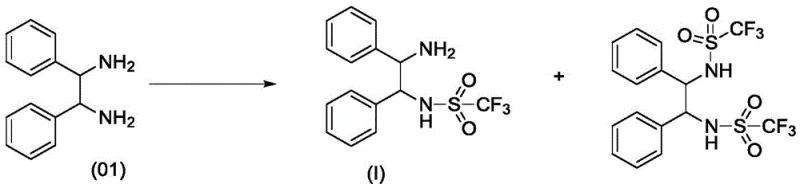
The Novel Approach
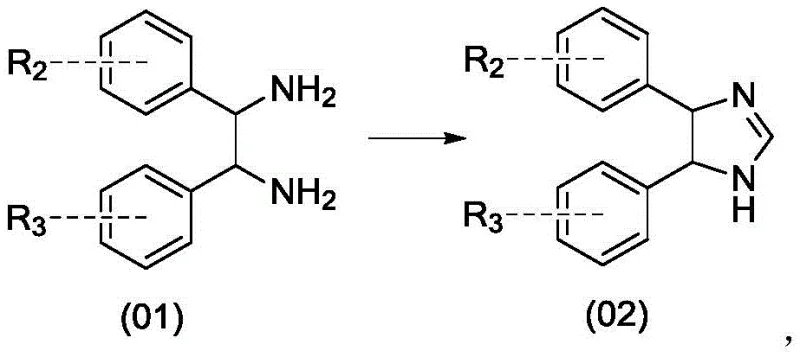
The innovative methodology disclosed in the patent circumvents these selectivity issues by employing a temporary masking strategy that fundamentally alters the reactivity of the substrate. Instead of attacking the diamine directly, the process first converts the 1,2-diamine into a 4,5-disubstituted-2-imidazoline derivative through condensation with N,N-dimethylformamide dimethyl acetal (DMF-DMA). This cyclization step effectively ties up both nitrogen atoms in a stable heterocyclic ring, preventing uncontrolled polysubstitution. Subsequent reaction with a sulfonyl anhydride occurs selectively at the imine nitrogen of the imidazoline ring, leading to a ring-opening event that generates the mono-substituted product with the second amine still protected as a formamide. This sequential approach ensures that only one nitrogen atom is functionalized with the desired leaving group, while the other remains masked until the final deprotection step. The result is a dramatic improvement in chemical purity, with disubstituted impurities reduced to trace levels below 0.5%, significantly simplifying the isolation and purification workflow.
Mechanistic Insights into Imidazoline-Mediated Selective Substitution
The core of this synthetic advancement lies in the unique reactivity of the imidazoline intermediate formed in the initial cyclization step. When the diamine reacts with DMF-DMA at elevated temperatures ranging from 65°C to 85°C, it undergoes a condensation reaction that eliminates methanol and forms a five-membered heterocyclic ring containing two nitrogen atoms. This structural modification is crucial because it breaks the symmetry of the original diamine and creates distinct electronic environments for the nitrogen atoms. The resulting imidazoline possesses a nucleophilic imine nitrogen that is more reactive towards electrophilic sulfonylating agents than the amine nitrogen would be in the open-chain form. By controlling the stoichiometry of the sulfonyl anhydride and maintaining low reaction temperatures between -30°C and 0°C, the reaction is kinetically driven to favor mono-substitution at the imine position. This selectivity is further enhanced by the steric environment of the cyclic intermediate, which hinders the approach of a second equivalent of the electrophile to the remaining nitrogen atom.
Following the selective substitution, the final stage involves the hydrolytic cleavage of the protecting group to reveal the free amine. The ring-opened intermediate, which contains a formamide moiety on the second nitrogen, is subjected to acidic or alkaline conditions to remove this group. Under acidic conditions, typically using hydrochloric acid concentrations of not less than 4 mol/L at temperatures around 80°C, the formamide bond is hydrolyzed to release the free amine and formic acid. Alternatively, alkaline hydrolysis using sodium hydroxide solutions can achieve the same result. This deprotection step is highly efficient and does not affect the newly installed sulfonyl group, ensuring that the final mono-substituted amine retains its structural integrity. The ability to tune the deprotection conditions allows manufacturers to optimize the process for specific substrates, ensuring maximum recovery of the product while minimizing degradation or racemization of the chiral centers. This mechanistic control is what enables the process to consistently deliver high-purity materials suitable for sensitive catalytic applications.
How to Synthesize Mono-Substituted Amine Efficiently
The implementation of this synthesis route requires careful attention to reaction parameters to maximize the benefits of the cyclization strategy. The process begins with the preparation of the imidazoline intermediate, followed by the controlled addition of the sulfonylating agent, and concludes with a straightforward hydrolytic workup. Operators must ensure strict temperature control during the substitution phase to prevent thermal degradation or side reactions, and the choice of solvent plays a pivotal role in the crystallization and isolation of the intermediates. The following guide outlines the standardized operational procedure derived from the patent examples, providing a clear roadmap for laboratory and pilot-scale execution. For detailed standard operating procedures and safety guidelines, please refer to the structured steps below.
- Cyclize the starting diamino compound (01) with N,N-dimethylformamide dimethyl acetal (DMF-DMA) at 40-100°C to form the imidazoline intermediate (02).
- React the imidazoline intermediate (02) with a sulfonyl anhydride (R1-O-R1) in the presence of a base at -30 to 0°C to perform ring-opening substitution, yielding the protected compound (03).
- Subject the protected compound (03) to acid or alkaline hydrolysis at 40-100°C to remove the protecting group and isolate the final mono-substituted amine (I).
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this patented methodology offers substantial advantages that directly impact the bottom line and supply chain stability for pharmaceutical manufacturers. By shifting from a non-selective direct substitution to a controlled cyclization-substitution sequence, companies can eliminate the costly and time-consuming purification steps associated with removing disubstituted impurities. The high selectivity of the reaction translates directly into higher effective yields, meaning less raw material is wasted in the form of unrecoverable byproducts. This efficiency gain is particularly significant when dealing with expensive chiral starting materials, where every percentage point of yield loss represents a significant financial burden. Furthermore, the robustness of the process reduces the risk of batch failures, ensuring a more predictable and reliable supply of critical intermediates for downstream drug synthesis.
- Cost Reduction in Manufacturing: The elimination of complex separation processes, such as preparative chromatography or multiple recrystallizations, leads to significant operational cost savings. By achieving high purity directly from the reaction mixture, manufacturers can reduce solvent consumption, energy usage for heating and cooling cycles, and labor hours required for purification. The use of common, inexpensive reagents like DMF-DMA and trifluoromethanesulfonic anhydride further contributes to a favorable cost profile compared to specialized chiral reagents. Additionally, the high yield of the cyclization step ensures that the expensive diamine starting material is utilized with maximum efficiency, minimizing the cost of goods sold for the final intermediate.
- Enhanced Supply Chain Reliability: The reliance on commercially available and stable reagents ensures that the supply chain is not vulnerable to the shortages often associated with exotic catalysts or custom-synthesized building blocks. The process operates under standard inert atmosphere conditions using common solvents like dichloromethane and ethanol, which are readily sourced from multiple global suppliers. This flexibility allows procurement teams to diversify their vendor base and negotiate better pricing, reducing the risk of production stoppages due to material scarcity. The scalability of the reaction, demonstrated from gram to multi-hundred gram scales in the patent examples, indicates a smooth path to tonnage production without the need for specialized equipment or hazardous high-pressure conditions.
- Scalability and Environmental Compliance: The process generates minimal hazardous waste compared to traditional methods that require heavy metal catalysts or generate large volumes of saline waste from excessive washing. The solvents used, such as ethyl acetate and methyl tert-butyl ether, are relatively environmentally benign and can be easily recovered and recycled through distillation. The high atom economy of the cyclization step, where the only byproduct is methanol, aligns with green chemistry principles and helps facilities meet increasingly stringent environmental regulations. This sustainability profile not only reduces waste disposal costs but also enhances the corporate social responsibility standing of the manufacturing site, making it a preferred partner for eco-conscious pharmaceutical clients.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this mono-substitution technology. These answers are derived directly from the experimental data and technical disclosures within the patent documentation, providing clarity on the feasibility and benefits of the route. Understanding these details is essential for R&D teams evaluating the technology for adoption and for supply chain managers assessing its impact on production planning. The responses highlight the specific advantages of the cyclization approach over legacy methods.
Q: Why is direct substitution of symmetric diamines problematic for pharmaceutical applications?
A: Direct substitution often leads to the formation of disubstituted impurities due to the symmetry of the diamine structure, making it difficult to isolate the desired mono-substituted product with high purity.
Q: How does the cyclization method described in CN110066233B improve product purity?
A: By temporarily cyclizing the diamine into an imidazoline ring, one amino group is effectively protected, forcing the subsequent substitution reaction to occur selectively at the remaining nitrogen, thereby drastically reducing disubstituted byproducts.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the process utilizes commercially available reagents like DMF-DMA and common solvents such as dichloromethane and ethanol, and operates under manageable temperature conditions, facilitating easy scale-up from kilogram to tonnage levels.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Mono-Substituted Amine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of high-purity intermediates in the development of next-generation pharmaceuticals. Our technical team has extensively analyzed the cyclization-based synthesis route described in CN110066233B and possesses the expertise to implement this technology at an industrial scale. We have extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory discovery to market supply is seamless and efficient. Our state-of-the-art facilities are equipped with rigorous QC labs capable of detecting impurities at trace levels, guaranteeing that every batch of mono-substituted amine meets stringent purity specifications required by global regulatory bodies. We are committed to delivering consistent quality and reliability for your most demanding chiral synthesis projects.
We invite you to collaborate with us to leverage this advanced synthetic methodology for your specific application needs. Our engineering team can provide a Customized Cost-Saving Analysis to quantify the potential economic benefits of switching to this high-yield process for your specific volume requirements. Please contact our technical procurement team today to request specific COA data from our pilot runs and to discuss route feasibility assessments tailored to your project timeline. Let us be your partner in transforming complex chemical challenges into commercial successes.