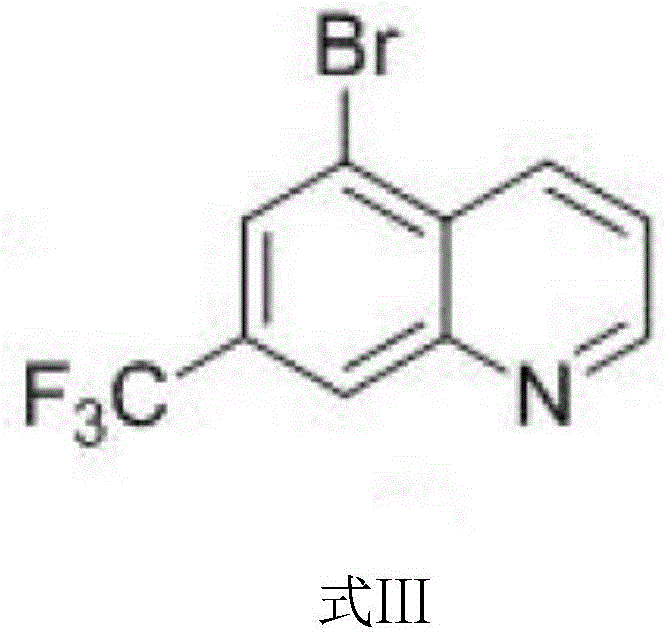
Advanced Synthesis of 5-Bromo-7-Trifluoromethyl Quinoline for Commercial Scale-Up and Drug Discovery
The pharmaceutical industry continuously seeks robust synthetic pathways for complex heterocyclic scaffolds, particularly quinoline derivatives which serve as critical backbones for kinase inhibitors. Patent CN108409649B introduces a transformative synthesis method for 5-bromo-7-trifluoromethyl quinoline, a high-value intermediate essential for developing antimalarial, antihypertensive, and anticancer agents targeting JNK and c-Met pathways. This technical breakthrough addresses long-standing challenges in regioselectivity and yield that have plagued previous manufacturing attempts. By shifting the bromination step to occur after the formation of the quinoline ring, the process eliminates the formation of inseparable isomers, thereby streamlining the purification workflow. For R&D directors and procurement specialists, this represents a significant opportunity to secure a reliable pharmaceutical intermediates supplier capable of delivering high-purity materials consistently. The strategic advantage lies not only in the chemical elegance but also in the operational simplicity, which translates directly into supply chain resilience and cost efficiency for downstream drug manufacturers.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of halogenated quinolines has been fraught with significant technical hurdles, primarily stemming from the lack of regiocontrol during the ring-forming step. Traditional approaches often attempt to introduce the bromine atom prior to cyclization, utilizing substrates like 3-bromo-5-trifluoromethylaniline in a Skraup condensation with glycerol. This legacy methodology invariably produces a complex mixture of isomers, specifically 5-bromo-7-trifluoromethylquinoline and its 7-bromo-5-trifluoromethyl counterpart, in comparable ratios. The structural similarity of these isomers makes separation via standard crystallization or distillation nearly impossible, often requiring expensive and low-yield chromatographic techniques that are impractical for commercial scale-up. Furthermore, the harsh acidic conditions required for these conventional condensations often lead to polymerization side reactions and tar formation, drastically reducing the overall mass balance and increasing waste disposal costs. For supply chain heads, these inefficiencies translate into unpredictable lead times and volatile pricing, as the effective yield of the desired isomer is often too low to sustain continuous manufacturing campaigns without significant batch-to-batch variability.
The Novel Approach
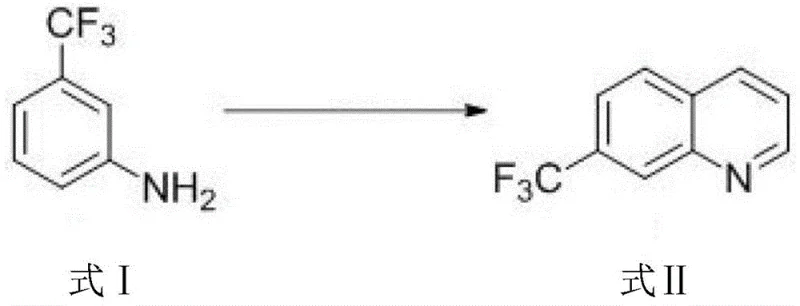
In stark contrast, the novel approach detailed in the patent data reverses the synthetic logic by prioritizing the construction of the quinoline core before introducing the halogen substituent. This strategy begins with the Skraup condensation of m-trifluoromethylaniline and glycerol to form 7-trifluoromethyl quinoline, a reaction that proceeds with high fidelity due to the electronic directing effects of the trifluoromethyl group. Once the stable quinoline scaffold is established, a regioselective bromination is performed using N-bromosuccinimide (NBS), which selectively targets the 5-position of the aromatic ring. This sequential methodology completely bypasses the isomerization issues inherent in the traditional route, ensuring that the crude reaction mixture is enriched with the target molecule. The operational benefits are profound, as the elimination of isomer separation steps simplifies the downstream processing unit operations significantly. This streamlined workflow not only enhances the total yield but also reduces the consumption of solvents and energy, aligning perfectly with modern green chemistry principles and cost reduction in pharmaceutical intermediates manufacturing objectives.
Mechanistic Insights into Skraup Condensation and Regioselective Bromination
The success of this synthesis relies heavily on the precise manipulation of electronic effects within the aromatic system during the cyclization phase. In the initial Skraup condensation, the electron-withdrawing nature of the meta-trifluoromethyl group on the aniline starting material influences the orientation of the glycerol-derived acrolein intermediate attack. Under the catalytic influence of sulfuric acid at elevated temperatures ranging from 120°C to 160°C, the dehydration and cyclization proceed to form the 7-trifluoromethyl substituted quinoline exclusively. This specificity is crucial because it sets the stage for the subsequent functionalization; the nitrogen atom in the newly formed quinoline ring exerts a strong deactivating effect on the pyridine ring, while the benzene ring remains more susceptible to electrophilic attack. Understanding this electronic distribution is vital for R&D teams aiming to replicate the process, as deviations in temperature or acid concentration could alter the kinetics of the cyclization, potentially leading to incomplete conversion or the formation of polymeric byproducts that complicate isolation.
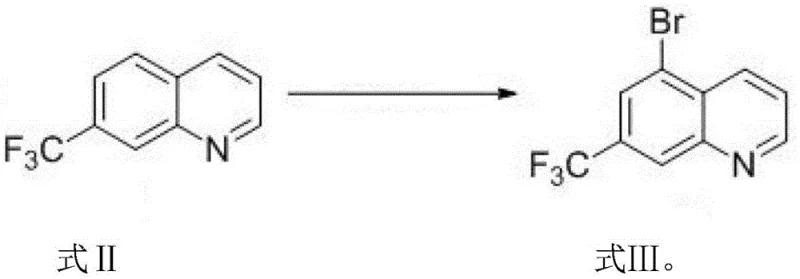
Following the ring closure, the bromination mechanism leverages the distinct reactivity profile of the 7-trifluoromethyl quinoline intermediate. When treated with N-bromosuccinimide in concentrated sulfuric acid, the generation of the electrophilic bromine species occurs in situ. The trifluoromethyl group at the 7-position and the ring nitrogen at the 1-position create a specific electronic environment that directs the incoming bromine electrophile to the 5-position. This regioselectivity is thermodynamically and kinetically favored over other positions on the benzene ring, such as the 6 or 8 positions, due to steric hindrance and resonance stabilization of the sigma complex intermediate. The reaction conditions, maintained between 60°C and 110°C for 3 to 5 hours, ensure complete conversion while minimizing the risk of polybromination or degradation of the sensitive trifluoromethyl moiety. This level of control over the impurity profile is what distinguishes this patent from prior art, offering a pathway to high-purity OLED material or pharmaceutical grade intermediates without the need for exhaustive recrystallization cycles.
How to Synthesize 5-Bromo-7-Trifluoromethyl Quinoline Efficiently
Implementing this synthesis route requires careful attention to reaction parameters to maximize the benefits of the patented methodology. The process is designed to be operationally simple, utilizing common industrial reagents such as glycerol, sulfuric acid, and NBS, which are readily available from global chemical suppliers. The initial condensation step demands precise temperature control to manage the exothermic nature of the reaction and ensure the complete dehydration of glycerol to the reactive enal species. Following the isolation of the quinoline intermediate, the bromination step must be conducted with adequate stirring and controlled addition of the brominating agent to maintain homogeneity and prevent local hot spots that could degrade the product. Detailed standardized synthesis steps see the guide below, which outlines the specific molar ratios, addition rates, and workup procedures necessary to achieve the reported high yields. Adhering to these protocols ensures that the commercial scale-up of complex pharmaceutical intermediates can be achieved with minimal technical risk and consistent quality output.
- Perform Skraup condensation on m-trifluoromethylaniline and glycerol in sulfuric acid at 120-160°C to form 7-trifluoromethyl quinoline.
- Conduct bromination reaction on the intermediate using N-bromosuccinimide (NBS) in concentrated sulfuric acid at 60-110°C.
- Quench the reaction, adjust pH to neutral, extract with ethyl acetate, and purify via column chromatography to obtain the final product.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this synthesis method offers substantial strategic advantages for organizations managing the procurement of specialized chemical building blocks. The primary value driver is the significant simplification of the purification process, which directly correlates to reduced manufacturing cycle times and lower operational expenditures. By eliminating the need to separate difficult isomers, the process reduces the consumption of chromatography media and organic solvents, which are often major cost centers in fine chemical production. This efficiency gain allows for a more predictable production schedule, enabling supply chain managers to plan inventory levels with greater confidence and reduce the safety stock required to buffer against production delays. Furthermore, the use of m-trifluoromethylaniline as a starting material leverages a commodity chemical with a stable global supply base, mitigating the risk of raw material shortages that often plague specialty syntheses reliant on exotic precursors.
- Cost Reduction in Manufacturing: The economic benefits of this route are derived principally from the enhanced atom economy and the drastic reduction in downstream processing requirements. Since the reaction avoids the formation of inseparable isomers, the yield loss associated with discarding off-spec material is virtually eliminated, leading to a higher effective output per batch. Additionally, the avoidance of transition metal catalysts in favor of organic reagents like NBS removes the necessity for expensive heavy metal scavenging steps, which are both costly and time-consuming. This streamlined approach results in substantial cost savings that can be passed down the supply chain, making the final API more competitive in the global market without compromising on quality standards.
- Enhanced Supply Chain Reliability: Supply continuity is critically dependent on the robustness of the synthetic route, and this method excels in its use of stable, non-hazardous reagents that are easy to transport and store. The reliance on sulfuric acid and glycerol, which are produced at massive scales globally, ensures that the manufacturing process is not vulnerable to the supply shocks that affect niche reagents. Moreover, the operational simplicity of the process means that it can be easily transferred between manufacturing sites or scaled up to larger reactor volumes without requiring specialized equipment or highly trained personnel. This flexibility significantly reduces lead time for high-purity pharmaceutical intermediates, allowing buyers to respond more agilely to fluctuations in market demand.
- Scalability and Environmental Compliance: As regulatory pressures regarding waste disposal and environmental impact intensify, the cleaner profile of this synthesis becomes a key differentiator. The reduction in solvent usage and the elimination of heavy metal contaminants simplify the wastewater treatment process, ensuring compliance with stringent environmental regulations in major manufacturing hubs. The high selectivity of the reaction minimizes the generation of hazardous byproducts, reducing the burden on waste management systems and lowering the overall environmental footprint of the production facility. This alignment with green chemistry principles not only mitigates regulatory risk but also enhances the corporate sustainability profile of the supply chain partners involved.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the patent specifications and are intended to clarify the feasibility of adopting this route for large-scale production. Understanding these details is essential for technical teams evaluating the integration of this intermediate into their existing drug development pipelines. The answers provided reflect the specific advantages of the patented method over legacy processes, focusing on yield, purity, and operational safety.
Q: Why is this synthesis method superior to conventional Skraup condensation routes?
A: Conventional methods often result in isomer mixtures that are difficult to separate, leading to low yields. This patented route avoids isomer formation by brominating after ring closure, ensuring high purity and simplified post-treatment.
Q: What are the key reaction conditions for the bromination step?
A: The bromination step utilizes N-bromosuccinimide (NBS) in concentrated sulfuric acid at temperatures between 60°C and 110°C for 3 to 5 hours, providing excellent regioselectivity at the 5-position.
Q: Is this process suitable for large-scale commercial production?
A: Yes, the process uses readily available raw materials like m-trifluoromethylaniline and glycerol, and avoids complex purification steps, making it highly scalable for industrial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 5-Bromo-7-Trifluoromethyl Quinoline Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the successful development of next-generation therapeutics. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can transition smoothly from laboratory discovery to full-scale manufacturing. We are committed to maintaining stringent purity specifications and operating rigorous QC labs to guarantee that every batch of 5-bromo-7-trifluoromethyl quinoline meets the exacting standards required for clinical and commercial applications. Our infrastructure is designed to support the complex chemistry involved in quinoline synthesis, providing you with a partner who understands the nuances of process safety and quality assurance.
We invite you to collaborate with us to optimize your supply chain and accelerate your time to market. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can support your long-term strategic goals. Let us help you secure a stable and cost-effective source of this vital pharmaceutical building block.