Advanced Synthesis of Dolutegravir Open-Loop Impurities for Global Pharmaceutical Quality Control
Advanced Synthesis of Dolutegravir Open-Loop Impurities for Global Pharmaceutical Quality Control
The pharmaceutical industry's relentless pursuit of safety and efficacy in antiretroviral therapies demands rigorous impurity profiling, particularly for complex molecules like Dolutegravir sodium. Patent CN110655517A introduces a groundbreaking preparation method for Dolutegravir open-loop impurities, specifically targeting Impurity A and Impurity B, which are critical degradation products formed during the shelf-life or stress testing of the bulk drug. Unlike traditional methods that rely on the unpredictable and low-yield degradation of the final active pharmaceutical ingredient (API), this invention establishes a dedicated, linear synthetic route starting from specialized pyridine derivatives. This approach not only solves the long-standing challenge of isolating these trace impurities from complex reaction mixtures but also provides a robust framework for generating high-purity reference standards essential for regulatory compliance and quality control laboratories worldwide.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the acquisition of specific degradation impurities for HIV integrase inhibitors has been a significant bottleneck in analytical method development. The conventional approach typically involves subjecting the Dolutegravir bulk drug to harsh stress conditions, such as refluxing in 6N hydrochloric acid, to force degradation. However, as detailed in the background of the patent, this brute-force method is fraught with inefficiencies; it yields only trace amounts of the target impurities alongside a myriad of other degradation byproducts. Separating these minute quantities of Impurity A and Impurity B from the complex matrix is technically arduous, often requiring preparative HPLC which is costly and difficult to scale. Furthermore, the instability of the side ring under acidic conditions means that the desired impurities can further degrade, leading to poor recovery rates and inconsistent batch-to-batch quality, thereby jeopardizing the reliability of the reference standards used for drug release testing.
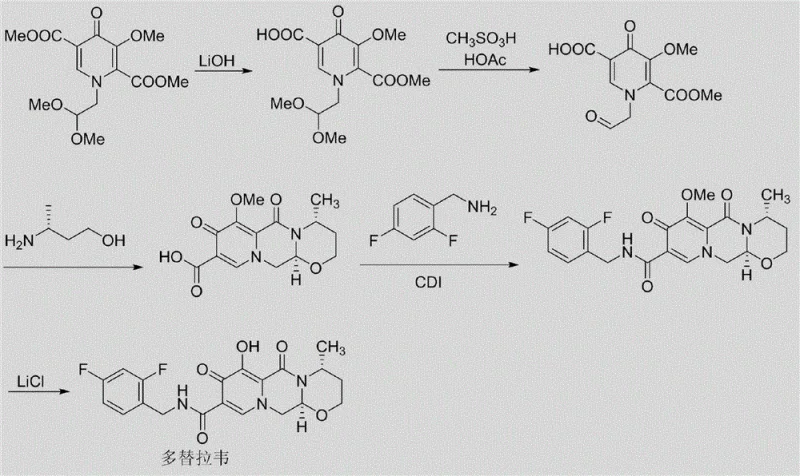
The Novel Approach
In stark contrast to the degradation-dependent strategies of the past, the methodology disclosed in CN110655517A employs a "bottom-up" synthetic design that constructs the impurity molecules from simpler precursors with high precision. This novel route utilizes a protection-ring closing-deprotection strategy that allows chemists to install the specific structural features of Impurity A and Impurity B in a controlled manner. By starting with Compound 1 and systematically building the molecular architecture through condensation, hydrolysis, and cyclization steps, the process avoids the chaotic mixture of byproducts associated with API degradation. This deliberate construction ensures that the target impurities are the major products of the reaction sequence, facilitating straightforward purification via crystallization or standard chromatography. The result is a scalable, reproducible process that delivers gram-to-kilogram quantities of high-purity impurities, effectively solving the supply dilemma for quality control teams.
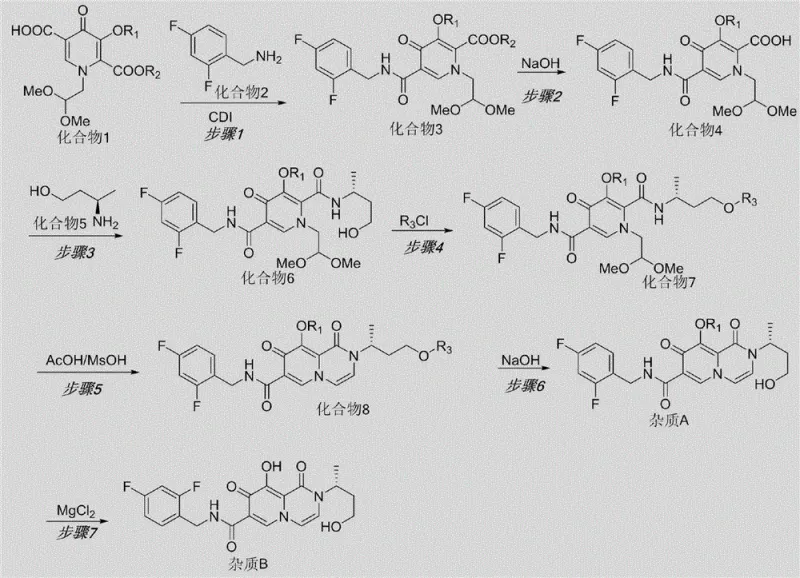
Mechanistic Insights into the Protection-Cyclization-Demethylation Strategy
The core innovation of this synthesis lies in its sophisticated handling of the sensitive oxazine ring system and the methoxy substituents. The formation of Impurity B, which represents a ring-opened and demethylated species, is mechanistically understood to arise from the instability of the side ring under acidic and thermal stress. The patent elucidates that the natural degradation pathway involves the hydrolysis of the ether linkage and subsequent ring opening. To mimic this synthetically without destroying the molecule, the inventors designed a route where the hydroxyl group is first protected (using benzoyl chloride) to prevent unwanted side reactions during the critical ring-closing step. The cyclization is then induced using a specific mixture of acetic acid and methanesulfonic acid, which provides the necessary protonation to close the ring while preserving the integrity of the rest of the scaffold. Finally, the strategic removal of the protecting group and the methoxy moiety using magnesium chloride allows for the precise generation of the phenolic hydroxyl group found in Impurity B, mirroring the natural degradation product with high fidelity.
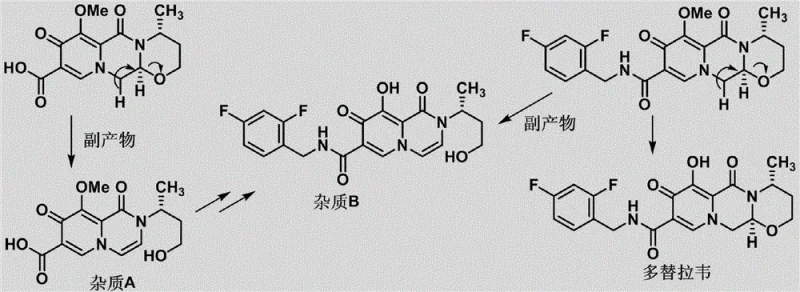
Furthermore, the control of stereochemistry and regioselectivity is paramount in this synthesis to ensure the impurities match those found in the actual drug product. The use of chiral starting materials, such as (R)-3-aminobutanol (Compound 5), ensures that the stereocenters in the final impurity structures are identical to those in Dolutegravir. The reaction conditions are meticulously tuned; for instance, the hydrolysis steps utilize mild alkaline conditions (1-2N NaOH) to selectively cleave ester bonds without affecting the amide linkages or the fluorophenyl groups. This selectivity is crucial because non-specific hydrolysis would lead to a loss of the difluorobenzyl carbamoyl side chain, generating irrelevant byproducts. The final demethylation step using MgCl2 is a gentle Lewis acid-mediated process that avoids the harsh conditions of traditional demethylating agents like BBr3, thereby minimizing the risk of over-reaction or decomposition of the sensitive pyridone core.
How to Synthesize Dolutegravir Impurities Efficiently
The synthesis of these critical reference standards follows a logical seven-step sequence that balances yield with operational simplicity. The process begins with the activation of the pyridine carboxylic acid derivative followed by amidation, setting the stage for the subsequent ring construction. Each step has been optimized for workup procedures that rely on standard aqueous washes and solvent exchanges, making the protocol highly adaptable for both laboratory and pilot plant environments. The detailed standardized synthesis steps, including specific molar ratios, solvent choices, and temperature controls for each transformation from Compound 1 to Impurity B, are outlined below to guide technical teams in replication.
- Condense Compound 1 with 2,4-difluorobenzylamine (Compound 2) using CDI to form the amide intermediate Compound 3.
- Hydrolyze the ester group of Compound 3 under alkaline conditions (NaOH) to yield the carboxylic acid Compound 4.
- React Compound 4 with (R)-3-aminobutanol (Compound 5) using EDC/HOBt coupling to generate the open-chain precursor Compound 6.
- Protect the hydroxyl group of Compound 6 with benzoyl chloride, followed by acid-catalyzed ring closure to form the bicyclic structure Compound 8.
- Deprotect Compound 8 to obtain Impurity A, then treat with Magnesium Chloride (MgCl2) to remove the methoxy group and yield Impurity B.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the shift from degradation-based isolation to dedicated synthesis represents a substantial opportunity for cost optimization and risk mitigation. The traditional reliance on degrading expensive API to obtain micrograms of impurity is inherently inefficient and creates a supply bottleneck; if the API production is delayed, the impurity standards cannot be made. This new method decouples the supply of impurities from the API manufacturing schedule, allowing for independent production planning. By utilizing commodity chemicals and standard reagents like CDI, EDC, and magnesium chloride, the raw material costs are drastically reduced compared to consuming the final high-value drug substance. Moreover, the ability to produce these impurities in large batches ensures a continuous supply for long-term stability studies and regulatory filings, eliminating the risk of stockouts that could delay product releases.
- Cost Reduction in Manufacturing: The elimination of the need to sacrifice valuable Dolutegravir API for impurity generation results in direct material savings. Additionally, the synthetic route avoids the use of exotic catalysts or extreme conditions that require specialized equipment, allowing production in standard glass-lined reactors. The high yields reported in the patent examples mean less solvent and energy are consumed per gram of product, further driving down the cost of goods sold (COGS) for these critical reference materials.
- Enhanced Supply Chain Reliability: Because the synthesis starts from stable, commercially available intermediates rather than the finished drug, the lead time for producing impurity standards is significantly shortened. This reliability is crucial for pharmaceutical companies managing tight regulatory timelines, as it ensures that analytical methods can be validated and stability protocols can be initiated without waiting for degradation samples to accumulate. The robustness of the chemistry also means that the process is less prone to failure, guaranteeing consistent delivery schedules.
- Scalability and Environmental Compliance: The reaction conditions described, such as room temperature condensations and ambient pressure filtrations, are inherently safer and easier to scale than high-pressure or cryogenic processes. The workup procedures primarily involve aqueous extractions and crystallizations, which generate less hazardous waste compared to complex chromatographic separations. This alignment with green chemistry principles simplifies environmental compliance and waste disposal, reducing the overall environmental footprint of the supply chain.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These answers are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on the feasibility and advantages of adopting this new methodology for impurity management.
Q: Why is a dedicated synthesis route for Dolutegravir impurities necessary compared to degradation studies?
A: Traditional degradation methods (e.g., acid reflux of the API) produce only trace amounts of impurities mixed with complex byproducts, making isolation and characterization extremely difficult. The patented route provides a dedicated, high-yield pathway to generate sufficient quantities of pure Impurity A and B for analytical reference standards.
Q: What are the critical reaction conditions for the ring-closing step in this synthesis?
A: The ring-closure step utilizes a mixed acid system of acetic acid and methanesulfonic acid in acetonitrile. This specific acidic environment facilitates the cyclization of the protected intermediate while maintaining the stability of the sensitive pyridone core, ensuring high conversion to the desired bicyclic structure.
Q: How does this method improve the supply chain for impurity reference standards?
A: By synthesizing impurities from readily available starting materials rather than relying on the degradation of the expensive final API, this method significantly reduces raw material costs and lead times. It allows for independent production scaling, ensuring a reliable supply of critical quality control materials regardless of API manufacturing fluctuations.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Dolutegravir Impurity Supplier
At NINGBO INNO PHARMCHEM, we understand that the integrity of your analytical data depends on the quality of your reference standards. Our team of expert chemists has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet your demand for Dolutegravir Impurity A and Impurity B with unmatched consistency. We operate stringent purity specifications and utilize rigorous QC labs equipped with state-of-the-art LC-MS and NMR instrumentation to verify the structural identity and purity of every batch, guaranteeing that our materials meet the exacting standards required by global regulatory agencies.
We invite you to collaborate with us to optimize your supply chain for HIV drug development. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our advanced synthesis capabilities can support your quality control objectives and accelerate your time to market.