Scalable Metal-Free Synthesis of Fevipiprant for Commercial API Production
The pharmaceutical industry is constantly seeking more efficient pathways to produce critical therapeutic agents, and the synthesis of Fevipiprant, a potent CRTh2 antagonist for asthma treatment, has seen a significant breakthrough with the publication of patent CN108484595B. This patent discloses a streamlined, four-step synthetic route that fundamentally alters the manufacturing landscape for this high-value active pharmaceutical ingredient (API). By shifting away from traditional transition-metal catalysis and complex protecting group strategies, this novel methodology offers a robust alternative that addresses long-standing issues regarding yield, purity, and environmental impact. For R&D directors and supply chain managers alike, understanding the nuances of this metal-free approach is essential for securing a reliable Fevipiprant supplier capable of meeting the rigorous demands of modern drug development.
Historically, the construction of the 7-azaindole core found in Fevipiprant has been a bottleneck in production. Conventional literature routes, such as those described in ACS Medicinal Chemistry Letters (2017), rely heavily on multi-step sequences involving benzenesulfonyl protective groups, methyl iodide alkylation, and the use of extremely strong bases like LDA and n-BuLi at cryogenic temperatures (-25°C). These conditions are not only hazardous and energy-intensive but also result in abysmal overall yields, reported to be as low as 2.1%. Furthermore, alternative routes utilizing Ullmann cyclization necessitate cuprous iodide, introducing the risk of heavy metal contamination that requires costly and time-consuming purification steps to meet regulatory limits.
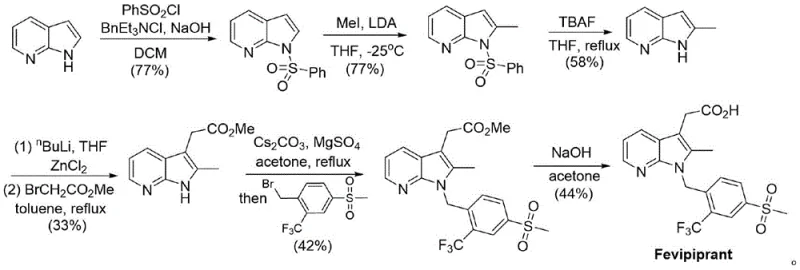
In stark contrast, the novel approach detailed in the patent utilizes a convergent strategy starting from readily available 1-(3-pyridyl)-2-acetone. The process begins with a mild oxidation to form a pyridine N-oxide, which serves as a key activated intermediate. This is followed by a direct cyclization with a functionalized benzyl amine using PyBrop (trispyrrolidinylphosphonium bromide hexafluorophosphate) as a coupling agent. This single step elegantly constructs the 7-azaindole scaffold while simultaneously installing the crucial side chain, bypassing the need for separate alkylation or protection steps. The subsequent conversion to the final acid involves a straightforward acylation-hydrolysis sequence and a final reduction, resulting in a process that is significantly shorter, safer, and more atom-economical than its predecessors.
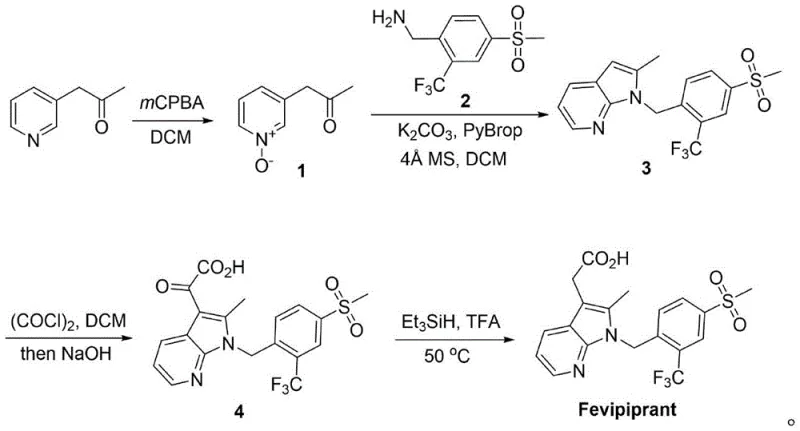
The mechanistic elegance of this synthesis lies in the activation of the pyridine ring via N-oxidation. In the key cyclization step (Step B), the N-oxide functionality increases the electrophilicity of the pyridine ring, facilitating nucleophilic attack by the amine component under the influence of the phosphonium salt PyBrop. This reaction proceeds under mild conditions (room temperature) in dichloromethane, avoiding the thermal stress and safety hazards associated with cryogenic organolithium chemistry. The subsequent transformation of the methyl group on the azaindole ring into the acetic acid side chain is achieved through a controlled acylation with oxalyl chloride followed by basic hydrolysis. Finally, the reduction of the resulting alpha-keto acid to the target acetic acid is performed using triethylsilane and trifluoroacetic acid, a method known for its chemoselectivity and ease of workup, ensuring high purity of the final API.
For procurement teams evaluating cost reduction in pharmaceutical intermediates manufacturing, the elimination of transition metals is a primary value driver. Traditional Ullmann-type couplings require stoichiometric or near-stoichiometric amounts of copper salts, which not only add to the raw material cost but also necessitate the use of specialized scavengers or chromatography to reduce residual metal levels below ppm thresholds mandated by health authorities. By adopting this metal-free protocol, manufacturers can drastically simplify the downstream processing workflow. The removal of copper scavenging steps translates directly into reduced solvent usage, lower waste disposal costs, and shorter batch cycle times, all of which contribute to substantial cost savings without compromising the quality of the final product.
From a supply chain perspective, the reliability of this route is enhanced by the use of stable, commercially available starting materials and reagents. The avoidance of air- and moisture-sensitive reagents like n-BuLi and LDA removes the need for specialized handling equipment and inert atmosphere protocols that can often lead to batch failures or delays. The reaction conditions are predominantly ambient or mildly heated (up to 50°C), making the process highly amenable to scale-up in standard glass-lined or stainless steel reactors. This operational simplicity ensures consistent batch-to-batch reproducibility, a critical factor for maintaining supply continuity for high-purity pharmaceutical intermediates. Furthermore, the high yields reported in the patent examples (e.g., 88% for the oxidation step and 82% for the final reduction) suggest a robust process capable of supporting commercial volume requirements.
The environmental profile of this synthesis is also markedly improved, aligning with the growing industry demand for green chemistry practices. The reduction in step count from lengthy multi-stage sequences to just four linear steps inherently reduces the cumulative E-factor (mass of waste per mass of product). By eliminating the benzenesulfonyl protecting group, the process avoids the generation of sulfonamide waste streams and the additional reagents required for deprotection. The use of common solvents like dichloromethane and ethyl acetate, while requiring proper management, allows for established recycling protocols. Overall, this methodology represents a significant advancement in the commercial scale-up of complex pharmaceutical intermediates, offering a cleaner, faster, and more economical path to market for Fevipiprant.
The commercial advantages of this patented synthesis extend beyond mere technical feasibility; they represent a strategic shift in how high-value asthma medications can be produced. By removing the dependency on scarce or expensive transition metal catalysts, the supply chain becomes more resilient to fluctuations in the commodities market. The simplified purification profile means that the time from reactor discharge to finished goods inventory is shortened, allowing for faster response to market demand. Additionally, the absence of genotoxic alkylating agents like methyl iodide, which are present in older routes, simplifies the toxicological assessment and regulatory filing process. These factors combined make this route an attractive option for generic manufacturers and innovators alike who are looking to optimize their production costs while maintaining stringent quality standards.
- Oxidize 1-(3-pyridyl)-2-acetone with mCPBA in DCM to form the pyridine N-oxide intermediate (Compound 1).
- Perform a PyBrop-mediated cyclization between Compound 1 and the trifluoromethyl-benzyl amine (Compound 2) to construct the 7-azaindole core (Compound 3).
- React Compound 3 with oxalyl chloride followed by hydrolysis to install the alpha-keto acid moiety, yielding Compound 4.
- Reduce the keto group of Compound 4 using triethylsilane and TFA to obtain the final Fevipiprant API.
Frequently Asked Questions (FAQ)
Q: How does this new synthesis route improve upon previous methods for Fevipiprant?
A: Unlike conventional routes that require transition metal catalysts (like Copper in Ullmann coupling) or harsh conditions (LDA/BuLi at -25°C), this patented method utilizes a metal-free PyBrop-mediated cyclization. This eliminates heavy metal contamination risks and simplifies purification, significantly boosting the total yield from roughly 2.1% in older literature to much higher practical yields.
Q: What are the key cost drivers reduced in this manufacturing process?
A: The process removes the need for expensive transition metal catalysts and their subsequent removal steps (scavengers/filtration). Additionally, it avoids the use of cryogenic conditions (-25°C) and protecting group strategies (benzenesulfonyl), which drastically reduces energy consumption and raw material costs associated with protection/deprotection cycles.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the route is designed for scalability. It operates under mild conditions (mostly room temperature to 50°C), uses common solvents like DCM, and involves only four linear steps. The absence of sensitive organometallic reagents makes the process robust and safer for tonnage-scale manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Fevipiprant Supplier
At NINGBO INNO PHARMCHEM, we understand that the transition from laboratory innovation to commercial reality requires a partner with deep technical expertise and scalable infrastructure. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the promising metrics of patent CN108484595B can be realized on an industrial level. We operate stringent purity specifications and maintain rigorous QC labs equipped to detect trace impurities, guaranteeing that every batch of Fevipiprant intermediate meets the highest global regulatory standards. Our commitment to quality assurance ensures that your supply chain remains uninterrupted and compliant.
We invite you to collaborate with us to leverage this advanced synthesis technology for your next project. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our optimized manufacturing processes can drive efficiency and profitability for your organization. Let us be your trusted partner in delivering high-quality pharmaceutical solutions to the market.