Revolutionizing Chiral Amide Synthesis: A Scalable Asymmetric C-H Arylation Strategy for Global Pharma Supply Chains
Introduction to Breakthrough Asymmetric C-H Activation Technology
The landscape of pharmaceutical intermediate manufacturing is undergoing a significant transformation driven by the need for more efficient and stereoselective synthetic methodologies. Patent CN116041216A introduces a groundbreaking method for the asymmetric C-H bond arylation of amide compounds, addressing critical challenges in the construction of chiral centers adjacent to carbonyl groups. This technology leverages a sophisticated palladium-catalyzed system that enables the direct functionalization of inert C(sp3)-H bonds with high precision. For R&D directors and process chemists, this represents a paradigm shift from traditional pre-functionalized coupling partners to direct C-H activation, streamlining synthetic routes for complex bioactive molecules. The ability to generate chiral amides with exceptional enantiomeric excess values opens new avenues for the rapid development of drug candidates, particularly those requiring specific stereochemical configurations for biological activity.
The significance of this invention extends beyond mere academic interest; it provides a practical solution for the industrial synthesis of high-value fine chemicals. Amide bonds are ubiquitous in medicinal chemistry, forming the backbone of countless therapeutic agents including peptides and peptidomimetics. By offering a method that operates under mild conditions with robust selectivity, this patent empowers reliable pharmaceutical intermediate suppliers to deliver higher quality materials with reduced process complexity. The integration of aryl fluoroborates as coupling partners further enhances the versatility of the reaction, allowing for the introduction of diverse aromatic motifs essential for optimizing the pharmacokinetic properties of final drug products.

The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the selective arylation of C(sp3)-H bonds in amides has been plagued by significant technical hurdles that hindered its widespread adoption in commercial manufacturing. Prior art, such as Rh(III)-catalyzed systems, often struggled with poor regioselectivity, leading to complex mixtures of isomers that were difficult and costly to separate. These conventional methods frequently required harsh reaction conditions, including elevated temperatures and strong oxidants, which could degrade sensitive functional groups present in advanced intermediates. Furthermore, the reliance on expensive rhodium catalysts without effective chiral control mechanisms resulted in low yields of the desired enantiomer, necessitating resource-intensive resolution steps. Such inefficiencies not only inflated the cost of goods but also extended development timelines, creating bottlenecks in the supply chain for critical active pharmaceutical ingredients.
The Novel Approach
In stark contrast, the methodology disclosed in CN116041216A offers a refined and highly efficient pathway that overcomes these longstanding deficiencies. By employing a tailored palladium catalyst system in conjunction with a novel chiral ligand derived from spiro-indane scaffolds, the invention achieves unprecedented levels of stereocontrol. The reaction proceeds smoothly at moderate temperatures ranging from 40°C to 50°C, preserving the integrity of delicate molecular architectures. This approach eliminates the need for pre-installed directing groups that require additional synthetic steps to install and remove, thereby shortening the overall synthetic sequence. The use of aryl fluoroborates as stable and easily handled coupling reagents further simplifies the operational protocol, making it highly attractive for cost reduction in API manufacturing where simplicity and reliability are paramount.
Mechanistic Insights into Pd-Catalyzed Asymmetric C-H Activation
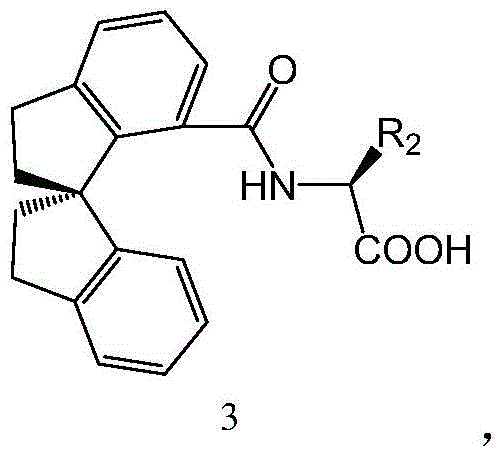
The core of this technological advancement lies in the intricate design of the chiral ligand, depicted as Formula 3 in the patent documentation. This ligand features a rigid spiro-indane backbone coupled with an amino acid moiety, creating a well-defined chiral pocket around the palladium center. This steric environment is crucial for differentiating between the enantiotopic C-H bonds during the activation step, ensuring that the arylation occurs with high fidelity. The mechanism likely involves a concerted metalation-deprotonation (CMD) pathway where the palladium species coordinates to the amide oxygen and activates the adjacent C-H bond. The presence of the chiral ligand dictates the facial selectivity of the subsequent migratory insertion of the aryl group from the fluoroborate species.
Furthermore, the reaction system incorporates a synergistic combination of additives that stabilize the catalytic cycle and promote turnover. Silver carbonate acts as a halide scavenger and facilitates the transmetallation step, while benzoquinone serves as a terminal oxidant to regenerate the active Pd(II) species from Pd(0). The inclusion of water as an additive is particularly noteworthy, as it can enhance the solubility of inorganic bases and facilitate proton transfer processes without compromising the anhydrous sensitivity typically associated with organometallic catalysis. This robust mechanistic framework ensures consistent performance across a broad substrate scope, providing R&D teams with a predictable tool for late-stage functionalization of complex molecules.
How to Synthesize Chiral Amides Efficiently
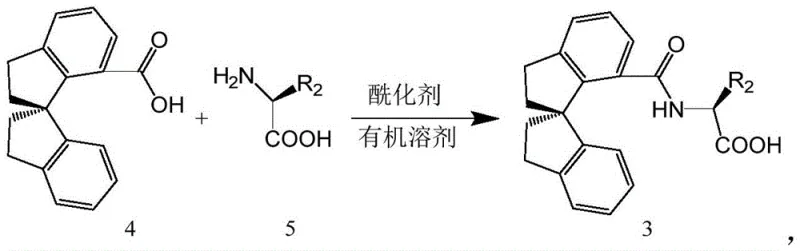
Implementing this asymmetric C-H arylation protocol requires careful attention to reagent quality and reaction parameters to maximize yield and enantioselectivity. The process begins with the preparation of the specific spiro-indane based ligand, which can be synthesized through a straightforward acylation of amino acids with spiro-indane carboxylic acids. Once the catalyst system is assembled, the reaction is conducted in tert-amyl alcohol, a solvent chosen for its ability to dissolve both organic substrates and inorganic additives while maintaining thermal stability. The detailed standardized synthesis steps for preparing the target chiral amides are outlined in the guide below, ensuring reproducibility for process scale-up.
- Prepare the reaction mixture by combining the amide substrate (Formula 2), aryl fluoroborate, Pd(OAc)2 catalyst, chiral spiro-indane ligand (Formula 3), Ag2CO3 additive, benzoquinone oxidant, Na2CO3 base, and water in tert-amyl alcohol solvent under nitrogen.
- Heat the reaction mixture to a mild temperature range of 40-50°C and stir continuously for 20-24 hours to ensure complete cross-coupling and high enantioselectivity.
- Upon completion, filter the reaction mixture to remove solid residues, wash the filtrate, and concentrate under reduced pressure to isolate the target chiral amide product (Formula 1) with high purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented technology translates into tangible strategic benefits that extend beyond the laboratory bench. The streamlined nature of the reaction directly addresses key pain points in chemical sourcing, such as the volatility of raw material costs and the complexity of multi-step syntheses. By reducing the number of unit operations required to install chiral aryl groups, manufacturers can significantly lower their operational overhead. The elimination of cryogenic conditions and the use of relatively inexpensive palladium salts instead of rarer metals contribute to a more resilient and cost-effective production model. This efficiency allows suppliers to offer more competitive pricing structures without compromising on the stringent quality standards required by the pharmaceutical industry.
- Cost Reduction in Manufacturing: The economic viability of this process is driven by its high atom economy and simplified workup procedures. Unlike traditional methods that may require multiple protection and deprotection steps or chromatographic separations to remove isomers, this direct C-H activation approach generates the target molecule with high purity directly from the reaction mixture. The use of commercially available aryl fluoroborates avoids the need for custom-synthesized organometallic reagents, which are often prone to instability and high logistics costs. Consequently, the overall cost of goods sold is optimized, enabling substantial cost savings in the production of high-value intermediates.
- Enhanced Supply Chain Reliability: Supply chain continuity is bolstered by the robustness of the reaction conditions and the availability of starting materials. The reagents involved, such as sodium carbonate and silver carbonate, are commodity chemicals with stable global supply networks, reducing the risk of shortages. Additionally, the mild reaction temperatures reduce energy consumption and equipment wear, leading to higher asset utilization rates. This reliability ensures that delivery schedules for critical pharmaceutical intermediates can be met consistently, mitigating the risk of production delays that could impact downstream drug formulation and market launch timelines.
- Scalability and Environmental Compliance: From an environmental and regulatory perspective, this method aligns well with green chemistry principles. The use of tert-amyl alcohol and water minimizes the generation of hazardous volatile organic compounds (VOCs) compared to processes relying on chlorinated solvents. The high selectivity of the reaction reduces the burden on waste treatment facilities by minimizing the volume of by-product streams. This environmental compatibility facilitates easier regulatory approval for manufacturing sites and supports corporate sustainability goals, making it an ideal choice for the commercial scale-up of complex pharmaceutical intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this asymmetric C-H arylation technology. These insights are derived directly from the experimental data and specifications provided in the patent literature, offering clarity on the practical application of the method. Understanding these details is essential for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios.
Q: What are the key advantages of this Pd-catalyzed method over traditional Rh-catalyzed C-H activation?
A: Unlike traditional Rh(III)-catalyzed methods which often suffer from poor regioselectivity and harsh conditions, this novel Pd-catalyzed protocol utilizes a specialized chiral spiro-indane ligand to achieve exceptional enantioselectivity (up to 99% ee) and high yields under mild temperatures (40-50°C), making it far more suitable for sensitive pharmaceutical intermediates.
Q: Is the chiral ligand used in this process commercially viable for large-scale production?
A: Yes, the ligand (Formula 3) is synthesized from readily available 7-carboxy-1,1'-spirodihydroindane precursors through a robust acylation process. The synthesis avoids exotic reagents, utilizing standard acylating agents like oxalyl chloride and common organic bases, ensuring a stable and cost-effective supply chain for the catalyst system.
Q: How does this technology impact the environmental footprint of amide manufacturing?
A: The process significantly reduces environmental impact by employing tert-amyl alcohol, a greener solvent, and incorporating water as a benign additive. Furthermore, the high selectivity minimizes the formation of by-products, reducing the need for extensive purification steps and lowering overall waste generation compared to conventional non-selective arylation methods.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Chiral Amides Supplier
At NINGBO INNO PHARMCHEM, we recognize the transformative potential of advanced catalytic technologies like the asymmetric C-H arylation described in CN116041216A. As a leading CDMO partner, we possess the technical expertise and infrastructure to translate such innovative laboratory protocols into robust commercial processes. Our team has extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the high selectivity and yield observed in the lab are maintained at an industrial scale. We operate stringent purity specifications and utilize rigorous QC labs to guarantee that every batch of chiral amide intermediate meets the exacting standards of global pharmaceutical clients.
We invite you to collaborate with us to leverage this cutting-edge chemistry for your next project. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific molecule, demonstrating how this route can optimize your budget. Please contact us to request specific COA data and route feasibility assessments, and let us help you secure a competitive advantage in the market with high-purity pharmaceutical intermediates delivered on time and within specification.