Advanced Synthesis of Ethyl 4-(1-hydroxyl-1-methylethyl)-2-propylimidazole-5-carboxylate for Commercial Scale-up
Advanced Synthesis of Ethyl 4-(1-hydroxyl-1-methylethyl)-2-propylimidazole-5-carboxylate for Commercial Scale-up
The pharmaceutical industry continuously seeks robust synthetic pathways for critical intermediates, particularly those serving as the backbone for blockbuster cardiovascular medications like Olmesartan. A pivotal advancement in this domain is detailed in patent CN102060778B, which discloses a highly efficient synthesis method for ethyl 4-(1-hydroxyl-1-methylethyl)-2-propylimidazole-5-carboxylate. This compound represents a sophisticated 2-substituted 4,5-dinitrile imidazole derivative that serves as a cornerstone in the construction of angiotensin II receptor blockers. The disclosed methodology addresses long-standing challenges in heterocyclic chemistry, specifically targeting the inefficiencies associated with traditional cyclization and functionalization steps. By leveraging a streamlined sequence that integrates imidate formation, ammonolysis, direct alcoholysis, and selective Grignard addition, the process achieves a remarkable total yield of 81%. This stands in stark contrast to historical benchmarks, offering a compelling value proposition for manufacturers aiming to optimize their supply chains for high-purity pharmaceutical intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of 2-substituted 4,5-dinitrile imidazoles has been plagued by operational complexities that hinder industrial scalability. Conventional literature methods often rely on acetonitrile as a solvent for the initial reflux reaction to generate the imidic ester intermediate. However, this approach necessitates a cumbersome solvent exchange step where acetonitrile must be distilled off and replaced with methanol or other solvents for the subsequent cyclization. This transition frequently results in the formation of difficult-to-handle residues where the product fails to precipitate as a solid, instead forming oily or viscous masses. Furthermore, the cyclization reaction in these older protocols is prone to generating significant quantities of polar, black viscous byproducts. These impurities are notoriously difficult to remove, as post-reaction decolorization with activated carbon proves ineffective once the dark polymeric substances have already formed. Consequently, these factors drastically reduce the overall yield to approximately 55% and compromise the physical quality of the crystals, making purification costly and time-consuming for procurement teams managing tight margins.
The Novel Approach
The innovative strategy outlined in the patent data fundamentally reengineers the process flow to eliminate these bottlenecks through solvent consistency and procedural reordering. Instead of switching solvents, the novel method utilizes a low-bo-point alcoholic solvent, such as methanol or ethanol, throughout the initial imidate formation and the subsequent cyclization steps. This continuity prevents the formation of stubborn residues and ensures a homogeneous reaction environment conducive to clean crystal growth. Crucially, the protocol advances the decolorization step; activated carbon is added directly to the reaction mixture during the initial reflux of the imidate precursor, rather than waiting until after cyclization. This proactive measure captures impurities early, preventing the formation of the problematic black viscous substances that characterize the old method. Additionally, the process merges the hydrolysis and esterification of the nitrile groups into a single direct alcoholysis step using hydrogen chloride in ethanol. This consolidation not only simplifies the operational workflow but also significantly enhances the material throughput, positioning this method as a superior choice for cost reduction in pharmaceutical intermediate manufacturing.
Mechanistic Insights into Imidazole Cyclization and Functionalization
The core of this synthetic breakthrough lies in the precise control of the cyclization mechanism and the subsequent functional group transformations. The reaction initiates with the condensation of diaminomaleonitrile and trimethyl orthobutyrate. In the presence of an alcoholic solvent under reflux conditions, these precursors undergo a rearrangement to form an imidic ester intermediate. The maintenance of the alcoholic environment is critical here, as it stabilizes the intermediate and facilitates the nucleophilic attack required for ring closure. Upon cooling the filtrate to a controlled temperature range of 0-15°C, the introduction of ammonia gas triggers the cyclization. The ammonia acts as a nucleophile, attacking the imidate carbon to close the imidazole ring, yielding 2-propylimidazole-4,5-dicarbonitrile. The low temperature is essential to control the exothermic nature of the ammonolysis and to promote the precipitation of the product as high-quality white crystals, minimizing the entrapment of mother liquor impurities.
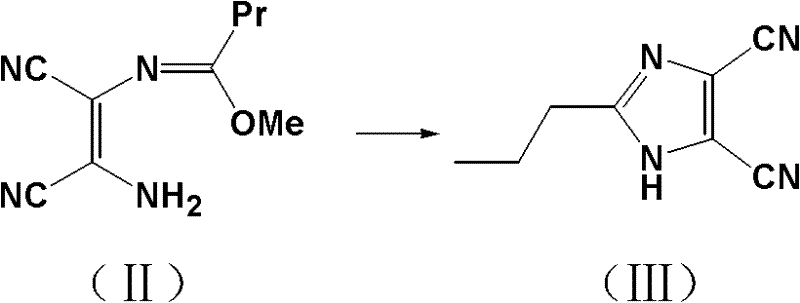
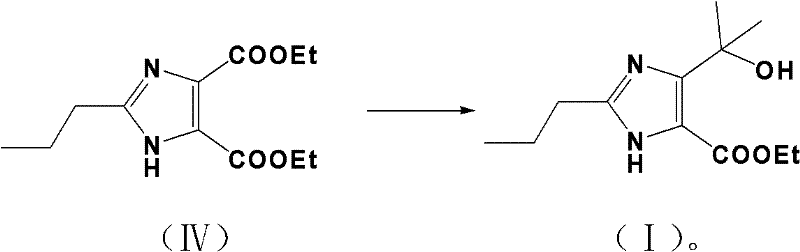
Following the formation of the dinitrile scaffold, the synthesis proceeds to the critical esterification and Grignard stages. The dinitrile is subjected to direct alcoholysis in an ethanol solution containing hydrogen chloride. This step effectively converts both nitrile groups into ethyl esters, yielding 2-propylimidazole-4,5-dicarboxylic acid diethyl ester. The final transformation involves a highly selective Grignard reaction. By reacting the diester with methylmagnesium bromide in an inert solvent like THF or ether at strictly controlled temperatures of 0-5°C, the process achieves mono-addition to one of the ester groups. This selectivity is paramount; it converts one ester moiety into a tertiary alcohol (the 1-hydroxyl-1-methylethyl group) while leaving the other ester intact. This precise chemoselectivity avoids the formation of di-alcohol byproducts, ensuring the final structure matches the target intermediate for Olmesartan with high fidelity.
How to Synthesize Ethyl 4-(1-hydroxyl-1-methylethyl)-2-propylimidazole-5-carboxylate Efficiently
Executing this synthesis requires strict adherence to the optimized parameters defined in the patent to ensure reproducibility and safety. The process begins with the preparation of the imidate intermediate, where the molar ratio of trimethyl orthobutyrate to diaminomaleonitrile is maintained between 1:1 and 1.1:1 to maximize conversion while minimizing excess reagent waste. The subsequent ammonolysis step demands careful thermal management, keeping the reaction mass between 0-15°C while introducing ammonia gas at a mass ratio of 1:1 relative to the starting diamine. For the esterification phase, the concentration of hydrogen chloride in the ethanol solution is a critical variable, typically ranging from 20% to 40% w/w, which dictates the reaction time from 2 to 7 days. Finally, the Grignard addition must be performed under an inert nitrogen atmosphere with a significant excess of the Grignard reagent (5:1 molar ratio) to drive the reaction to completion without compromising selectivity. Detailed standardized operating procedures for these steps are essential for technology transfer.
- Reflux trimethyl orthobutyrate and diaminomaleonitrile in an alcoholic solvent with activated carbon to form the imidate intermediate, followed by hot filtration.
- Cool the filtrate to 0-15°C and introduce ammonia gas to cyclize and precipitate 2-propylimidazole-4,5-dicarbonitrile.
- Perform direct alcoholysis of the dinitrile using hydrogen chloride in ethanol under reflux to obtain the diethyl ester derivative.
- React the diethyl ester with methylmagnesium bromide in THF or ether at 0-5°C to selectively form the final hydroxy-ethyl ester product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route offers tangible strategic benefits that extend beyond simple yield metrics. The primary advantage lies in the drastic simplification of the unit operations. By eliminating the need for solvent swapping between the imidate formation and cyclization steps, the process reduces the consumption of energy and solvents, directly lowering the variable costs associated with manufacturing. Furthermore, the early integration of activated carbon treatment ensures that the crude product is of higher quality before it even reaches the crystallization phase. This reduces the burden on downstream purification equipment and minimizes the loss of valuable material during filtration and washing, leading to substantial cost savings in the overall production budget. The robustness of the crystallization steps also means that the process is less sensitive to minor fluctuations in operating conditions, enhancing batch-to-batch consistency.
- Cost Reduction in Manufacturing: The elimination of complex solvent exchange procedures and the consolidation of hydrolysis and esterification into a single step significantly reduce processing time and utility consumption. By avoiding the formation of intractable black viscous byproducts, the need for extensive rework or specialized purification techniques is removed, streamlining the production line and optimizing resource utilization for high-volume manufacturing.
- Enhanced Supply Chain Reliability: The raw materials utilized in this process, such as diaminomaleonitrile and trimethyl orthobutyrate, are commercially available commodity chemicals with stable supply chains. The high overall yield of 81% ensures that less raw material is required to produce the same amount of finished goods, reducing the strain on upstream suppliers and mitigating the risk of shortages. This efficiency translates to more reliable delivery schedules for downstream API manufacturers.
- Scalability and Environmental Compliance: The process relies on standard chemical engineering operations like reflux, filtration, and crystallization, which are easily scalable from pilot plants to multi-ton reactors. The use of alcoholic solvents and the avoidance of hazardous heavy metal catalysts or exotic reagents simplify waste treatment protocols. This alignment with green chemistry principles facilitates easier regulatory approval and reduces the environmental footprint of the manufacturing facility.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing a clear understanding of the process capabilities. Understanding these details is crucial for R&D teams evaluating the feasibility of adopting this route for their own production lines or for procurement specialists assessing the quality standards of potential suppliers.
Q: What is the total yield advantage of this new synthesis method compared to literature?
A: The patented method achieves a total yield of 81%, which is significantly superior to the approximately 55% yield reported in existing literature methods that suffer from difficult solid precipitation and poor decolorization.
Q: How does this process improve impurity control during the cyclization step?
A: By advancing the activated carbon decolorization step to the initial imidate formation stage and maintaining a consistent alcoholic solvent system, the process effectively removes polar black viscous byproducts before cyclization, resulting in white crystalline products with purity exceeding 99%.
Q: Is this synthesis route suitable for large-scale industrial production?
A: Yes, the process eliminates complex solvent swaps and utilizes standard reflux and crystallization operations. The use of readily available raw materials like diaminomaleonitrile and the high overall yield make it highly scalable for commercial manufacturing.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Ethyl 4-(1-hydroxyl-1-methylethyl)-2-propylimidazole-5-carboxylate Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-quality intermediates play in the development of life-saving cardiovascular therapies. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated chemistry described in patent CN102060778B can be translated into reliable industrial reality. We maintain stringent purity specifications and operate rigorous QC labs equipped with advanced analytical instrumentation to verify every batch against the highest international standards. Our commitment to excellence ensures that the impurity profiles of our intermediates are tightly controlled, providing our partners with the confidence needed to navigate complex regulatory filings.
We invite global pharmaceutical partners to collaborate with us to leverage this advanced synthesis technology for your supply chain. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to reach out today to obtain specific COA data and comprehensive route feasibility assessments, allowing you to make informed decisions that drive efficiency and innovation in your drug development pipeline.