Advanced Manufacturing of Hepatitis B Virus Nucleocapsid Inhibitor Intermediates via Copper-Catalyzed Cyclization
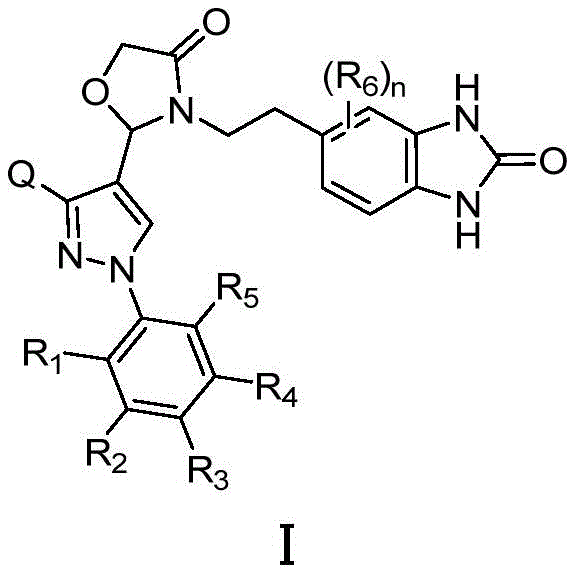
The pharmaceutical industry is constantly seeking more efficient and safer pathways to synthesize complex active pharmaceutical ingredients (APIs) and their critical intermediates. Patent CN115677681A discloses a groundbreaking preparation method for a hepatitis B virus nucleocapsid inhibitor, specifically targeting the compound of Formula I. This innovation represents a significant leap forward in medicinal chemistry, addressing the urgent need for functional cures for chronic hepatitis B, a condition where current market drugs often fall short of achieving complete viral clearance. The disclosed technology focuses on optimizing the synthetic route to not only enhance yield and product purity but also to drastically reduce the presence of genotoxic impurities, a critical parameter for regulatory approval and patient safety.
The core of this invention lies in a novel cyclization strategy that bypasses traditional, hazardous nitration steps. By utilizing a copper-catalyzed ring-closure reaction on a specifically designed urea intermediate, the process achieves a robust and scalable manufacturing protocol. This approach is particularly relevant for reliable pharmaceutical intermediate suppliers looking to modernize their production capabilities. The structural complexity of Formula I, featuring a fused benzimidazolone system linked to a pyrazole core, demands precise control over stereochemistry and regioselectivity, which this new method delivers with exceptional consistency.
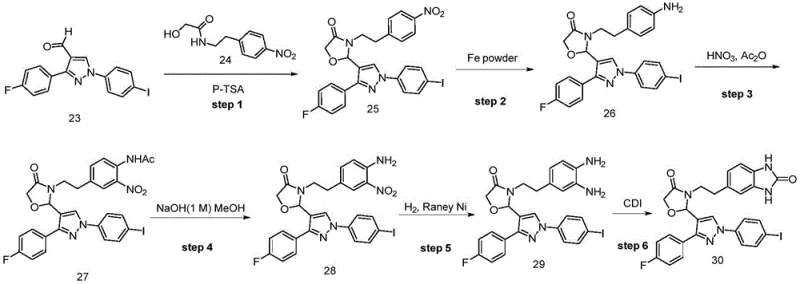
Prior to this innovation, the synthesis of similar benzimidazolone derivatives often relied on cumbersome multi-step sequences involving high-risk reagents. As detailed in the background of the patent, conventional methods such as those described in WO2017173999 A1 typically involve a nitration reaction using concentrated nitric acid and acetic anhydride. This traditional approach presents severe limitations for industrial application. The nitration step is highly exothermic, posing significant safety risks regarding thermal runaway and potential multiple nitrations, which complicates process control and increases the likelihood of dangerous incidents during commercial scale-up of complex pharmaceutical intermediates.
Furthermore, the legacy synthetic routes suffer from poor overall yields due to the accumulation of losses across multiple purification steps. Specifically, the final ring-closure in older methods often requires four distinct steps from key intermediates, with individual yields dropping as low as 24.9% in the final stage. Crucially, these older processes frequently necessitate silica gel column chromatography for purification. While acceptable for gram-scale laboratory synthesis, column chromatography is economically and logistically prohibitive for metric-ton manufacturing, creating a bottleneck for cost reduction in API manufacturing. Additionally, intermediates in these traditional routes often contain nitrobenzene or o-phenylenediamine motifs, which are flagged as structural alerts for genotoxicity, requiring expensive and rigorous ppm-level analytical controls to ensure patient safety.
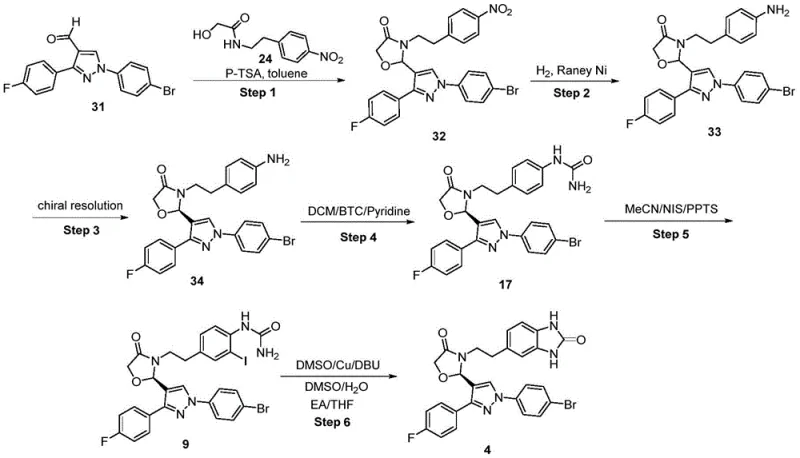
In stark contrast, the novel approach disclosed in CN115677681A offers a streamlined, safer, and more efficient pathway. The new method initiates with the formation of a urea linkage from an aniline precursor, followed by a selective halogenation and a final copper-catalyzed cyclization. This sequence effectively eliminates the need for hazardous nitration reactions entirely. The optimization of the reaction conditions allows for the direct formation of the benzimidazolone core with significantly higher efficiency. For instance, the total yield from the chiral intermediate to the final product in the new route can reach up to 65.9%, a substantial improvement over the roughly 44.6% total yield observed in comparative prior art examples. This increase in efficiency directly translates to reduced raw material consumption and waste generation.
The mechanistic heart of this innovation is the copper-catalyzed intramolecular coupling reaction, akin to an Ullmann-type condensation. In this critical step, a halogenated urea intermediate (Formula II) undergoes cyclization to form the target benzimidazolone ring (Formula I). The patent specifies a wide range of effective copper catalysts, including copper powder, cuprous iodide, cuprous chloride, and various copper salts, often used in conjunction with specific ligands such as 1,10-phenanthroline derivatives or oxalamides like L19 and L21. The choice of base is also pivotal, with strong organic bases like DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) proving highly effective in promoting the reaction in polar aprotic solvents like DMSO.
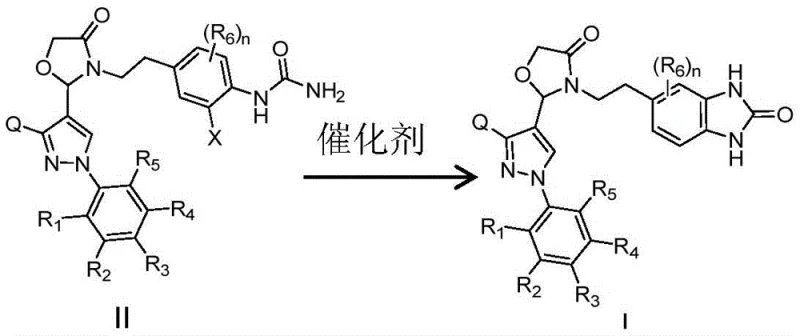
From an impurity control perspective, this mechanistic pathway offers distinct advantages. By avoiding the formation of nitro-groups and primary aromatic amines in the late stages of synthesis, the process inherently minimizes the risk of generating genotoxic impurities. The intermediates generated in the final three steps of this new route have been confirmed to be negative in Ames tests, providing a much cleaner safety profile. This simplifies the quality control burden, as there is no need to develop and validate ultra-sensitive analytical methods to track trace levels of mutagenic byproducts. The ability to achieve high purity (often exceeding 99%) through simple crystallization and filtration, rather than chromatography, further ensures that the final product meets the stringent specifications required for clinical and commercial use.
How to Synthesize Hepatitis B Inhibitor Intermediate Efficiently
The synthesis of these high-value intermediates relies on a carefully orchestrated sequence of reactions that balance reactivity with selectivity. The process begins with the conversion of an aniline derivative into a urea using a carbonylation reagent like triphosgene, followed by immediate reaction with ammonia. This urea is then selectively halogenated at the ortho-position relative to the urea nitrogen using reagents such as N-iodosuccinimide (NIS). The final and most critical step involves the copper-mediated cyclization. Detailed operational parameters, including specific temperature ranges (typically 60-120°C for cyclization), solvent choices, and catalyst loading ratios, are essential for maximizing yield and minimizing side reactions. For a comprehensive guide on executing this synthesis with precision, please refer to the standardized protocol below.
- React the aniline precursor with a carbonylation reagent such as triphosgene to form an isocyanate intermediate, followed by immediate amination to generate the urea derivative.
- Perform selective halogenation on the urea intermediate using agents like N-iodosuccinimide (NIS) or N-bromosuccinimide (NBS) to introduce the necessary halogen atom for cyclization.
- Execute the final ring-closing reaction using a copper catalyst system (e.g., copper powder or CuI with DBU) in a polar aprotic solvent like DMSO to form the benzimidazolone core.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthetic route offers transformative benefits that extend beyond mere chemical elegance. The primary advantage lies in the drastic simplification of the manufacturing process, which directly impacts the cost of goods sold (COGS). By eliminating the need for column chromatography, the process removes a major cost driver associated with silica gel consumption, solvent usage, and labor-intensive batch processing. This shift to crystallization-based purification enables continuous or large-batch processing, significantly enhancing throughput and reducing the time required to bring products to market.
- Cost Reduction in Manufacturing: The elimination of hazardous nitration steps reduces the need for specialized corrosion-resistant equipment and extensive safety infrastructure, leading to lower capital expenditure (CAPEX) and operational expenditure (OPEX). Furthermore, the higher overall yield means that less starting material is required to produce the same amount of final product, optimizing raw material utilization. The avoidance of expensive and difficult-to-remove genotoxic impurities also reduces the cost associated with analytical testing and waste disposal, contributing to substantial cost savings throughout the product lifecycle.
- Enhanced Supply Chain Reliability: The reliance on readily available and stable reagents, such as copper powders and common organic bases, ensures a robust supply chain that is less susceptible to disruptions compared to routes requiring specialized or hazardous reagents like concentrated nitric acid. The milder reaction conditions and improved safety profile facilitate easier transportation and storage of intermediates, reducing logistical complexities. This reliability is crucial for maintaining consistent supply to downstream API manufacturers and ensuring uninterrupted drug production.
- Scalability and Environmental Compliance: The process is inherently designed for scalability, moving seamlessly from kilogram-scale development to multi-ton commercial production without the bottlenecks associated with chromatographic purification. The reduction in hazardous waste, particularly the avoidance of nitration byproducts and silica gel waste, aligns with green chemistry principles and simplifies environmental compliance. This makes the process not only economically attractive but also environmentally sustainable, meeting the increasing regulatory and corporate social responsibility demands of the global pharmaceutical industry.
Frequently Asked Questions (FAQ)
Understanding the technical nuances of this new synthesis method is vital for stakeholders evaluating its potential for integration into their supply chains. The following questions address common inquiries regarding the safety, scalability, and regulatory implications of this technology. These answers are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering a clear picture of the operational realities.
Q: How does this new process improve safety compared to traditional nitration routes?
A: The novel method eliminates the use of concentrated nitric acid and acetic anhydride required in traditional nitration steps, significantly reducing the risk of thermal runaway and exothermic hazards associated with large-scale production.
Q: What are the purity advantages of avoiding column chromatography?
A: By optimizing reaction conditions and crystallization protocols, the process achieves high purity (over 99%) through simple filtration and washing, removing the need for silica gel column chromatography which is costly and difficult to scale.
Q: Does this synthesis route address genotoxicity concerns?
A: Yes, the intermediates in the final three steps of this optimized route have tested negative for genotoxicity in Ames assays, unlike previous routes that involved nitrobenzene or o-phenylenediamine structures, ensuring better compliance with ICH M7 guidelines.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Hepatitis B Inhibitor Intermediate Supplier
The technological advancements detailed in CN115677681A highlight the critical importance of having a manufacturing partner who can translate complex laboratory innovations into reliable commercial reality. NINGBO INNO PHARMCHEM stands at the forefront of this capability, possessing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the specific requirements of copper-catalyzed reactions and sensitive crystallization processes, ensuring that every batch meets stringent purity specifications. With our rigorous QC labs and commitment to quality, we guarantee that the intermediates supplied are free from critical genotoxic impurities and ready for the next stage of API synthesis.
We invite pharmaceutical companies and research institutions to collaborate with us to leverage this optimized synthetic route for your hepatitis B inhibitor programs. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this new process can lower your overall production costs. Please contact our technical procurement team today to request specific COA data, route feasibility assessments, and samples to evaluate the superior quality of our intermediates. Let us be your partner in bringing safer and more effective hepatitis B treatments to patients worldwide.