Advanced Manufacturing of Latricinib Intermediate via Chiral Induction Technology
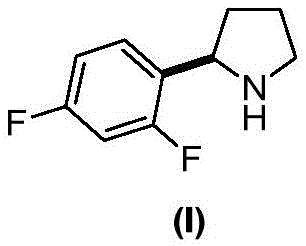
The rapid advancement of oncology therapeutics has placed significant demand on the supply chain for high-purity kinase inhibitors, particularly Tropomyosin Receptor Kinase (TRK) inhibitors like Latricinib. As the pharmaceutical industry seeks to optimize the manufacturing of these critical drugs, the efficiency of intermediate synthesis becomes a paramount concern for both cost and regulatory compliance. A pivotal development in this sector is detailed in Chinese Patent CN107445879B, which discloses a streamlined preparation method for the key chiral intermediate (R)-2-(2,5-difluorophenyl)pyrrolidine. This patent represents a strategic shift away from complex, multi-step organometallic sequences toward a more robust chiral induction pathway. By leveraging a chiral Betti base derivative, the disclosed method achieves high stereocontrol while drastically simplifying the operational parameters required for production. For global procurement teams and R&D directors, understanding this technological pivot is essential for securing a reliable pharmaceutical intermediates supplier capable of meeting the rigorous standards of modern drug development.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of chiral pyrrolidine intermediates for TRK inhibitors has relied heavily on sophisticated and often hazardous organometallic chemistry. As illustrated in previous international patents such as WO2010/048314, traditional routes frequently employ chiral ligands like sparteine alongside reactive species such as butyllithium and zinc chloride. These processes typically necessitate cryogenic conditions, often dropping temperatures to -78°C, to maintain control over reactivity and stereoselectivity. Furthermore, the reliance on palladium-catalyzed cross-coupling reactions introduces significant challenges regarding heavy metal removal, a critical quality attribute for any Active Pharmaceutical Ingredient (API). The requirement for strictly anhydrous and anaerobic environments not only increases capital expenditure for specialized equipment but also complicates the scale-up process, making cost reduction in API manufacturing difficult to achieve. These factors collectively create a bottleneck in the supply chain, leading to longer lead times and higher vulnerability to production disruptions.
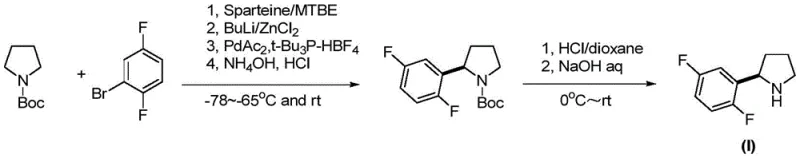
The Novel Approach
In stark contrast to the convoluted pathways of the past, the methodology described in CN107445879B introduces a elegant solution centered on nucleophilic substitution and reduction. This approach utilizes a chiral induction reagent, specifically a benzotriazole-substituted oxazine, which acts as a robust scaffold for introducing the difluorophenyl group. The reaction sequence begins with a Grignard substitution, followed by a straightforward reduction and a final debenzylation step. This strategy effectively bypasses the need for transition metal catalysts in the bond-forming steps, thereby eliminating the associated purification burdens. The operational window is significantly widened, with reaction temperatures ranging from -30°C to ambient conditions, which is far more manageable than the extreme cryogenics of prior art. This simplification translates directly into enhanced process safety and operational controllability, making it an ideal candidate for the commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Chiral Induction and Grignard Substitution
The core of this synthetic innovation lies in the precise manipulation of stereochemistry through the chiral Betti base framework. The process initiates with the reaction of (7aR,10R,12S)-10-(1-benzotriazolyl)-12-phenyl-7a,8,9,10-tetrahydro-12H-naphtho[1,2-e]pyrrolo[2,1-b][1,3]oxazine with 2,5-difluorophenylmagnesium bromide. In this step, the benzotriazole moiety serves as an excellent leaving group, facilitating a clean nucleophilic attack by the Grignard reagent at the C-10 position. The steric environment provided by the fused ring system ensures that the incoming aryl group adopts the desired orientation, preserving the optical purity established in the starting material. Following this substitution, the intermediate undergoes reduction using lithium aluminum hydride. This powerful reducing agent selectively cleaves the oxazine ring and reduces the iminium functionality without affecting the sensitive fluorine substituents on the aromatic ring. The result is a naphthol derivative where the chiral center is firmly established, ready for the final liberation of the pyrrolidine ring.
Controlling impurities in chiral synthesis is often the most challenging aspect for R&D teams, yet this route offers inherent advantages in this regard. By avoiding palladium catalysts, the risk of generating genotoxic impurities or persistent metal residues is virtually eliminated, simplifying the analytical profile of the final product. The use of a stoichiometric Grignard reagent allows for precise control over the reaction kinetics, minimizing the formation of homocoupling byproducts that are common in catalytic cross-couplings. Furthermore, the final debenzylation step can be achieved through catalytic hydrogenation using palladium on carbon or via oxidative cleavage with ceric ammonium nitrate. This flexibility allows manufacturers to choose the method that best aligns with their existing infrastructure and waste management capabilities. The overall atom economy is improved by reducing the number of discrete isolation steps, ensuring that the high-purity pharmaceutical intermediates produced meet the stringent specifications required for oncology drug formulations.
How to Synthesize (R)-2-(2,5-difluorophenyl)pyrrolidine Efficiently
Implementing this synthesis route requires careful attention to reagent quality and temperature control, although the conditions are markedly milder than traditional methods. The process is designed to be linear and scalable, starting from the chiral oxazine precursor and proceeding through substitution and reduction to the final deprotected amine. Operators should ensure that the Grignard reagent is freshly prepared or titrated to ensure accurate stoichiometry, as this directly impacts the yield of the substitution step. The reduction phase requires careful quenching to manage the exotherm associated with lithium aluminum hydride, but standard safety protocols for hydride reductions are sufficient. For the final step, catalytic hydrogenation is generally preferred for its cleanliness, though oxidative methods provide a viable alternative if hydrogenation infrastructure is limited. The detailed standardized synthesis steps see the guide below for specific operational parameters.
- Perform a nucleophilic substitution reaction between a chiral benzotriazole-containing oxazine and 2,5-difluorophenylmagnesium bromide at controlled low temperatures.
- Execute a reduction reaction on the substitution product using lithium aluminum hydride to generate the naphthol derivative intermediate.
- Conduct a final debenzylation reaction via catalytic hydrogenation or ceric ammonium nitrate oxidation to yield the target pyrrolidine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this novel synthesis route offers compelling economic and logistical benefits that extend beyond simple yield improvements. The elimination of noble metal catalysts such as palladium acetate and complex ligands like tri-n-butylphosphonium tetrafluoroborate removes a significant cost driver from the bill of materials. Moreover, the avoidance of cryogenic temperatures reduces energy consumption and allows for the use of standard glass-lined reactors rather than specialized low-temperature vessels. This shift fundamentally alters the cost structure of production, enabling substantial cost savings that can be passed down the supply chain. Additionally, the robustness of the Grignard substitution reaction enhances supply chain reliability by reducing the sensitivity to minor fluctuations in reaction conditions, thereby minimizing batch failures and ensuring consistent delivery schedules for critical drug substances.
- Cost Reduction in Manufacturing: The most immediate financial impact comes from the removal of expensive transition metal catalysts and the simplification of the purification workflow. Traditional methods often require extensive chromatography or scavenging steps to remove trace metals to ppb levels, which adds significant time and material costs. By utilizing a metal-free bond formation strategy, this new process drastically reduces the burden on downstream processing. The reagents employed, such as 2,5-difluorophenylmagnesium bromide and lithium aluminum hydride, are commodity chemicals available from multiple global suppliers, fostering a competitive pricing environment. This diversification of raw material sources mitigates the risk of supply shortages and price volatility, ensuring a more stable cost basis for long-term manufacturing contracts.
- Enhanced Supply Chain Reliability: Operational simplicity is a key driver of supply chain resilience. The ability to run reactions at temperatures between -30°C and ambient conditions, rather than -78°C, means that production is less susceptible to cooling system failures or seasonal variations in utility availability. The reduced sensitivity to moisture and oxygen compared to organolithium-based routes further decreases the likelihood of batch rejection due to environmental excursions. This robustness allows for tighter production planning and shorter cycle times, effectively reducing lead time for high-purity pharmaceutical intermediates. Manufacturers can maintain higher inventory turnover rates and respond more agilely to fluctuations in market demand for TRK inhibitors, providing a strategic advantage in a competitive therapeutic landscape.
- Scalability and Environmental Compliance: From an environmental and regulatory perspective, this process aligns well with green chemistry principles. The reduction in heavy metal usage lowers the toxicity profile of the waste stream, simplifying effluent treatment and disposal compliance. The solvent systems utilized, such as tetrahydrofuran and methanol, are well-understood and easily recoverable, supporting sustainable manufacturing practices. The linear nature of the synthesis, with fewer intermediate isolations, reduces the overall solvent footprint and energy intensity per kilogram of product. These factors make the process highly attractive for commercial scale-up, as it minimizes the environmental permitting hurdles often associated with complex organometallic chemistry. Consequently, this route supports the long-term sustainability goals of pharmaceutical companies while ensuring uninterrupted supply of vital oncology medications.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of Latricinib intermediates using this patented technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on the practical implementation of the method. Understanding these details is crucial for technical teams evaluating the feasibility of adopting this route for their own manufacturing pipelines or for sourcing partners who utilize this technology.
Q: What are the primary advantages of this new synthesis route over prior art methods?
A: The novel method eliminates the need for expensive noble metal catalysts like palladium and harsh organolithium reagents, significantly simplifying the purification process and reducing heavy metal residue risks in the final API.
Q: How does the chiral induction strategy ensure stereochemical purity?
A: By utilizing a pre-formed chiral Betti base derivative as the starting scaffold, the stereochemistry is locked in early, avoiding the need for difficult resolution steps later in the synthesis.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process utilizes readily available Grignard reagents and standard reduction conditions, avoiding extreme cryogenic temperatures and inert atmosphere requirements that hinder scalability.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable (R)-2-(2,5-difluorophenyl)pyrrolidine Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of advanced oncology therapies depends on a secure and high-quality supply of critical intermediates. Our technical team has extensively analyzed the pathway described in CN107445879B and possesses the expertise to execute this chiral induction strategy with precision. We bring extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project can move seamlessly from clinical trials to full-scale market launch. Our facilities are equipped with state-of-the-art rigorous QC labs capable of verifying stringent purity specifications, including chiral HPLC analysis to guarantee enantiomeric excess. We are committed to delivering a reliable pharmaceutical intermediates supplier experience that prioritizes both quality and continuity of supply.
We invite you to engage with our technical procurement team to discuss how this optimized synthesis route can benefit your specific project requirements. By leveraging our manufacturing capabilities, you can access a Customized Cost-Saving Analysis tailored to your volume needs, demonstrating the tangible economic advantages of this metal-free approach. We encourage potential partners to contact us directly to request specific COA data and route feasibility assessments, allowing you to make informed decisions based on hard data. Together, we can accelerate the development of life-saving TRK inhibitors by ensuring a robust and efficient supply chain for this vital intermediate.