Scalable Production of High-Purity Chiral Alpha-Difluoromethylamine Building Blocks
Introduction to Advanced Fluorinated Building Blocks
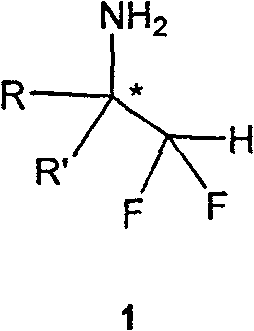
The integration of fluorine atoms into organic molecules has revolutionized modern medicinal chemistry, particularly through the strategic placement of the difluoromethyl (CF2H) moiety. As detailed in Chinese Patent CN100560561C, the development of efficient routes to optically pure α-difluoromethylamines represents a critical advancement for the pharmaceutical industry. These chiral compounds serve as indispensable building blocks for the synthesis of complex bioactive agents, including enzyme inhibitors and peptidomimetics. The unique electronic properties of the CF2H group allow it to function as a lipophilic hydrogen bond donor and a robust bioisostere for the hydroxymethyl group, thereby improving the metabolic stability and membrane permeability of drug candidates without compromising biological activity.
Historically, accessing these structures with high stereochemical fidelity has been a formidable challenge for process chemists. Traditional approaches often suffered from poor enantioselectivity or required harsh conditions incompatible with sensitive functional groups. The methodology disclosed in the patent overcomes these limitations by employing a nucleophilic asymmetric difluoromethylation strategy. This approach leverages the reactivity of difluoromethyl phenyl sulfone in the presence of strong non-nucleophilic bases to attack chiral sulfinyl imines. The result is a robust, scalable process capable of delivering target amines with exceptional optical purity, addressing a long-standing gap in the supply of high-quality fluorinated intermediates for global drug development pipelines.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this innovation, the synthetic landscape for chiral α-difluoromethylamines was fraught with inefficiencies and technical barriers. Early attempts relied heavily on the resolution of racemic mixtures or the use of chiral auxiliaries that offered limited stereocontrol. For instance, palladium-catalyzed asymmetric hydrogenation of difluoromethyl imidates, while conceptually attractive, frequently resulted in disappointingly low enantiomeric excess (ee) values, often hovering around 30%. Such poor selectivity necessitates extensive and wasteful downstream purification processes, such as repeated recrystallization or preparative chiral HPLC, which drastically inflate manufacturing costs and reduce overall throughput.
Furthermore, alternative strategies involving the reduction of chiral β-sulfinyl-N-aryl imines or the use of L-proline catalyzed Mannich reactions exhibited significant substrate scope limitations. These methods often displayed slow reaction kinetics and were sensitive to steric hindrance, failing to accommodate a diverse range of aromatic and aliphatic substituents. Additionally, multi-step sequences starting from difluoroacetates or the use of Freon-22 derivatives introduced unnecessary complexity and safety hazards. The cumulative effect of these drawbacks was a lack of a general, reliable method for producing optically pure materials, creating a bottleneck for R&D teams aiming to explore the full potential of fluorinated amine pharmacophores in new drug entities.
The Novel Approach
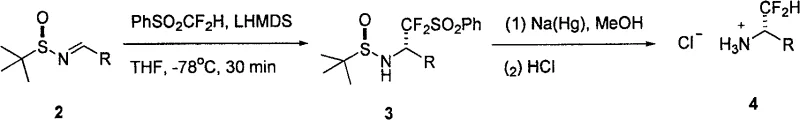
The patented methodology introduces a paradigm shift by utilizing difluoromethyl phenyl sulfone (PhSO2CF2H) as a stable and readily available nucleophilic source of the CF2H group. In this streamlined process, the sulfone reagent reacts with (R)- or (S)-tert-butylsulfinyl aldimines under the influence of a strong base such as lithium hexamethyldisilazide (LHMDS). This reaction proceeds with remarkable efficiency at low temperatures, typically around -78°C, to yield the corresponding N-sulfinyl sulfonamide intermediates with diastereomeric ratios exceeding 99:1. The chiral information encoded in the tert-butylsulfinyl group effectively directs the facial selectivity of the nucleophilic attack, ensuring the formation of a single stereoisomer.
Following the initial coupling, the process employs a mild reductive cleavage protocol to remove both the phenylsulfonyl and tert-butylsulfinyl protecting groups. This can be achieved using sodium amalgam or sodium metal in methanol, followed by acidification to isolate the final amine as a stable hydrochloride salt. This two-step sequence—addition followed by deprotection—is operationally simple and avoids the use of toxic transition metals or hazardous gases. The versatility of this approach is evidenced by its compatibility with a wide array of substrates, including electron-rich and electron-deficient aryl rings, heterocycles like furan, and various alkyl chains, making it a universal solution for the synthesis of diverse fluorinated amine libraries.
Mechanistic Insights into Asymmetric Nucleophilic Difluoromethylation
The success of this synthetic route hinges on the precise interplay between the electrophilic chiral imine and the nucleophilic difluoromethyl carbanion generated in situ. When LHMDS deprotonates the difluoromethyl phenyl sulfone, it creates a stabilized carbanion species that is sufficiently nucleophilic to attack the imine carbon but controlled enough to prevent side reactions. The tert-butylsulfinyl group attached to the imine nitrogen plays a dual role: it activates the imine towards nucleophilic addition through coordination with the lithium cation, and it imposes a rigid steric environment that blocks one face of the planar imine bond. This chelation-controlled transition state ensures that the incoming CF2H group attacks exclusively from the less hindered face, locking in the desired stereochemistry with high fidelity.
From an impurity control perspective, this mechanism offers distinct advantages over radical-based or transition-metal catalyzed pathways. Because the reaction does not involve free radical intermediates or redox cycles that can generate complex byproduct profiles, the crude reaction mixture is remarkably clean. The primary impurities are typically unreacted starting materials or minor diastereomers that can be easily removed during the workup or the subsequent crystallization of the hydrochloride salt. The stability of the sulfone reagent also minimizes the formation of decomposition products often seen with more labile difluorocarbene precursors. This inherent cleanliness simplifies the purification workflow, reducing solvent consumption and waste generation, which aligns perfectly with the principles of green chemistry and sustainable manufacturing.
How to Synthesize Optically Pure Alpha-Difluoromethylamine Efficiently
Implementing this synthesis in a laboratory or pilot plant setting requires careful attention to temperature control and reagent stoichiometry to maximize yield and stereoselectivity. The process begins with the preparation of the chiral imine substrate, which is reacted with the sulfone reagent in an anhydrous solvent like tetrahydrofuran (THF) under an inert atmosphere. The addition of the base must be performed slowly at cryogenic temperatures to manage the exotherm and maintain the integrity of the reactive intermediates. Following the coupling reaction, the mixture is warmed to room temperature to ensure complete conversion before quenching. The subsequent deprotection step involves treating the intermediate with a reducing agent in an alcoholic solvent, followed by acidification to precipitate the product.
- React chiral tert-butylsulfinyl aldimine with difluoromethyl phenyl sulfone and LHMDS in THF at -78°C to form the sulfonamide intermediate.
- Perform reductive desulfonylation using sodium amalgam or sodium in methanol to remove the phenylsulfonyl group.
- Treat the resulting amine with hydrochloric acid to obtain the final optically pure alpha-difluoromethylamine hydrochloride salt.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented technology translates into tangible operational benefits and risk mitigation. The elimination of precious transition metal catalysts, such as palladium, removes a significant cost driver from the bill of materials. More importantly, it obviates the need for specialized and expensive metal scavenging resins or rigorous analytical testing for heavy metal residues, which are critical regulatory hurdles in API manufacturing. By relying on abundant organic reagents and standard inorganic bases, the supply chain becomes more resilient to fluctuations in the market prices of rare earth metals or catalysts, ensuring consistent production costs and reliable availability of raw materials.
Furthermore, the high stereoselectivity of the process directly impacts the cost of goods sold by minimizing material loss during purification. Achieving greater than 99% ee at the reaction stage means that fewer batches need to be reprocessed or discarded due to failing chiral purity specifications. This efficiency shortens the overall production cycle time and increases the effective capacity of existing manufacturing assets. The robustness of the chemistry also facilitates easier technology transfer from R&D to commercial scale, as the reaction parameters are well-defined and tolerant of minor variations. This reliability reduces the lead time for scaling up new fluorinated intermediates, allowing pharmaceutical partners to accelerate their clinical timelines and bring life-saving therapies to market faster.
- Cost Reduction in Manufacturing: The process utilizes cost-effective reagents like difluoromethyl phenyl sulfone and avoids the use of expensive chiral transition metal complexes. This fundamental shift in reagent selection significantly lowers the direct material costs associated with producing high-value fluorinated intermediates. Additionally, the simplified workup procedures reduce the consumption of solvents and chromatography media, further driving down operational expenses.
- Enhanced Supply Chain Reliability: By depending on commercially available and stable starting materials, manufacturers can secure long-term supply contracts with reduced risk of shortage. The absence of specialized catalysts that may have long lead times or single-source dependencies enhances the overall security of supply. This stability is crucial for maintaining continuous production schedules for key pharmaceutical intermediates.
- Scalability and Environmental Compliance: The reaction conditions are amenable to large-scale batch processing, with straightforward temperature and mixing requirements. The avoidance of toxic heavy metals simplifies waste treatment and disposal, ensuring compliance with stringent environmental regulations. This eco-friendly profile supports corporate sustainability goals and reduces the regulatory burden associated with hazardous waste management.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of these specialized fluorinated compounds. Understanding the nuances of this synthesis helps stakeholders make informed decisions about integrating these building blocks into their development pipelines. The answers provided are derived directly from the experimental data and technical specifications outlined in the patent literature, ensuring accuracy and relevance for industrial applications.
Q: What is the optical purity achievable with this difluoromethylation method?
A: The patented process consistently achieves optical purity (ee values) greater than 99%, significantly outperforming traditional palladium-catalyzed hydrogenation methods which often yield less than 30% ee.
Q: Why is the CF2H group valuable in drug design?
A: The difluoromethyl group acts as a lipophilic hydrogen bond donor and serves as an isoelectric and isopolar bioisostere for the hydroxymethyl (-CH2OH) group, enhancing metabolic stability and membrane permeability in drug candidates.
Q: Does this synthesis require expensive transition metal catalysts?
A: No, this method utilizes organolithium bases like LHMDS rather than precious transition metals like palladium, eliminating the need for costly metal scavenging steps and reducing heavy metal impurities in the final API.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Alpha-Difluoromethylamine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical role that high-purity fluorinated intermediates play in the discovery and development of next-generation therapeutics. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the demands of both early-stage research and late-stage clinical supply. We adhere to stringent purity specifications and operate rigorous QC labs equipped with advanced chiral HPLC and NMR capabilities to guarantee that every batch of optically pure α-difluoromethylamine meets the highest quality standards required by global regulatory agencies.
We invite you to collaborate with us to leverage this advanced synthetic technology for your specific project needs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your volume requirements and timeline. Please contact us to request specific COA data for our catalog compounds or to discuss route feasibility assessments for custom targets. By partnering with us, you gain access to a reliable supply chain partner committed to delivering excellence in fine chemical manufacturing and innovation.