Advanced Synthesis of 1-Trifluoromethyl-Tetra-Substituted Cyclopentene Derivatives for Pharmaceutical Applications
The pharmaceutical and fine chemical industries are constantly seeking robust methodologies to access fluorinated carbocyclic scaffolds, which are pivotal in modern drug design due to their enhanced metabolic stability and lipophilicity. Patent CN107324982B discloses a groundbreaking preparation method for 1-trifluoromethyl-tetra-substituted cyclopentene derivatives, addressing a significant gap in the synthetic availability of these valuable motifs. This technology leverages a novel cyclization strategy that transforms readily available 2-trifluoromethyl-1,3-conjugated eneyne compounds and 1,3-dicarbonyl compounds into highly functionalized cyclopentene cores. The significance of this invention lies not only in its ability to construct complex quaternary carbon centers but also in its operational simplicity, making it an attractive candidate for the reliable pharmaceutical intermediate supplier looking to diversify their portfolio with fluorinated building blocks.
The structural versatility of the resulting compounds, as exemplified by the general formula (III), allows for extensive downstream derivatization, which is crucial for medicinal chemistry campaigns aiming to optimize lead compounds. By introducing the trifluoromethyl group directly onto the cyclopentene ring, chemists can impart desirable physicochemical properties to potential drug candidates without requiring late-stage fluorination steps that are often hazardous or low-yielding. This patent represents a strategic advancement in the field of organofluorine chemistry, providing a direct route to skeletons found in various bioactive molecules.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of trifluoromethyl-substituted cyclopentenes has been fraught with challenges that hinder their widespread adoption in industrial settings. Traditional routes often rely on multi-step sequences involving harsh reaction conditions, such as high temperatures or strong acidic environments, which can compromise sensitive functional groups present in complex molecular architectures. Furthermore, many existing methods suffer from poor regioselectivity and stereoselectivity, leading to difficult-to-separate mixtures of isomers that drastically reduce the overall process efficiency. The use of expensive or toxic reagents in conventional protocols also raises significant environmental and safety concerns, complicating the waste management processes required for commercial scale-up of complex pharmaceutical intermediates.
Another critical bottleneck in prior art is the limited substrate scope, where minor changes in the electronic or steric properties of the starting materials can lead to complete reaction failure or negligible yields. This lack of generality forces process chemists to develop bespoke synthetic routes for each new analog, consuming valuable time and resources during the drug development timeline. Additionally, the difficulty in introducing the trifluoromethyl group at the desired position with high fidelity often necessitates the use of specialized fluorinating agents that are costly and require stringent handling procedures, thereby inflating the cost reduction in fine chemical manufacturing efforts.
The Novel Approach
In stark contrast to these legacy issues, the methodology described in CN107324982B offers a streamlined, one-pot cyclization process that operates under remarkably mild conditions. By utilizing a silver-catalyzed system, this novel approach enables the direct coupling of conjugated enynes with 1,3-dicarbonyl compounds to form the target cyclopentene ring with high efficiency. The reaction proceeds at room temperature, eliminating the need for energy-intensive heating or cooling systems, which is a substantial advantage for green chemistry initiatives. The tolerance for a broad spectrum of functional groups, including esters, nitriles, nitro groups, and halogens, demonstrates the robustness of this catalytic system, allowing for the rapid generation of diverse chemical libraries.
The operational simplicity of this new route cannot be overstated; it utilizes commercially available starting materials and standard laboratory solvents like toluene, removing the dependency on exotic reagents. The workup procedure is equally straightforward, involving basic filtration and chromatographic purification, which facilitates easier technology transfer from the laboratory to pilot plant scales. This method effectively bypasses the need for pre-functionalized cyclic precursors, constructing the ring and installing the trifluoromethyl group simultaneously, thereby shortening the synthetic linear step count and improving the overall atom economy of the process.
Mechanistic Insights into Silver-Catalyzed Cyclization
The core of this transformative synthesis lies in the activation of the alkyne moiety within the 2-trifluoromethyl-1,3-conjugated eneyne substrate by the silver catalyst. Silver ions, acting as a soft Lewis acid, coordinate with the pi-system of the triple bond, increasing its electrophilicity and rendering it susceptible to nucleophilic attack by the active methylene group of the 1,3-dicarbonyl compound. This initial interaction triggers a cascade of intramolecular rearrangements and cyclization events that ultimately forge the five-membered carbocyclic ring. The presence of the trifluoromethyl group plays a crucial electronic role, stabilizing intermediate species through inductive effects while ensuring the final product retains the desired fluorination pattern essential for biological activity.
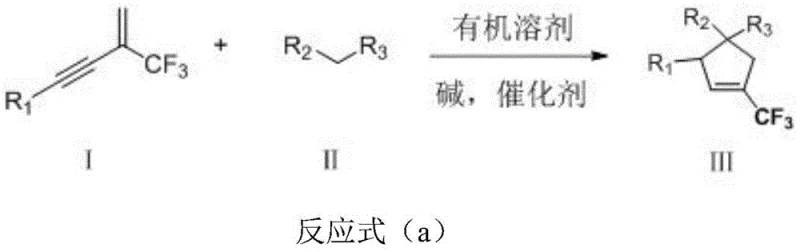
The choice of base, typically triethylamine, is critical for deprotonating the 1,3-dicarbonyl compound to generate the necessary nucleophile without decomposing the sensitive silver catalyst or the fluorinated substrate. The reaction mechanism likely proceeds through a silver-acetylide or silver-alkyne complex intermediate, which directs the regioselectivity of the ring closure to favor the formation of the 1-trifluoromethyl-tetra-substituted isomer over other potential regioisomers. Understanding this mechanistic pathway allows process chemists to fine-tune reaction parameters, such as catalyst loading and solvent polarity, to maximize yield and purity. The use of dried toluene as the solvent further ensures that moisture-sensitive intermediates are protected, preventing hydrolysis side reactions that could degrade the product quality.
Impurity control is inherently built into this mechanism due to the high specificity of the silver-catalyzed cyclization. Unlike radical-based fluorination methods that often produce a plethora of poly-fluorinated byproducts, this ionic pathway is highly selective for the mono-trifluoromethylated cyclopentene structure. The mild reaction temperature of 25°C minimizes thermal degradation of the reactants and products, ensuring a cleaner crude profile that simplifies downstream purification. This level of control over the impurity profile is vital for meeting the stringent purity specifications required for active pharmaceutical ingredients, reducing the burden on analytical teams to identify and quantify trace contaminants.
How to Synthesize 1-Trifluoromethyl-Tetra-Substituted Cyclopentene Efficiently
To implement this synthesis effectively, one must adhere to the optimized protocol detailed in the patent examples, which balances reagent stoichiometry with reaction time to achieve optimal conversion. The process begins with the careful preparation of the reaction environment, ensuring an inert atmosphere to protect the silver catalyst from oxidation or precipitation. The precise addition of the base and the order of reagent mixing are key operational details that influence the reaction kinetics and final yield. Following the standardized procedure ensures reproducibility across different batches, which is a fundamental requirement for any reliable pharmaceutical intermediate supplier aiming to deliver consistent quality to their clients.
- Dissolve the 2-trifluoromethyl-1,3-conjugated enyne compound and silver nitrate catalyst in dried toluene under a nitrogen atmosphere.
- Add triethylamine and the 1,3-dicarbonyl compound to the reaction mixture at room temperature.
- Stir the reaction for 24 hours, monitor by TLC, then filter through diatomaceous earth and purify via column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, this technology offers compelling advantages driven by the accessibility and cost-effectiveness of the raw materials. The starting enynes and 1,3-dicarbonyl compounds are commodity chemicals that can be sourced from multiple vendors globally, mitigating the risk of supply chain disruptions associated with single-source specialty reagents. The elimination of expensive noble metal catalysts like palladium or rhodium in favor of silver nitrate represents a significant cost optimization, as silver salts are generally more affordable and easier to recover or dispose of responsibly. This shift in catalyst selection directly contributes to cost reduction in fine chemical manufacturing by lowering the bill of materials for each production run.
The mild reaction conditions translate into substantial energy savings and enhanced operational safety, which are critical factors for supply chain reliability. Operating at room temperature removes the need for complex heating or cryogenic cooling infrastructure, allowing the process to be run in standard glass-lined reactors without specialized modifications. This simplicity facilitates faster batch turnover times and reduces the likelihood of thermal runaway incidents, ensuring a steady and predictable supply of intermediates. Furthermore, the straightforward workup procedure involving filtration and evaporation minimizes solvent consumption and waste generation, aligning with increasingly strict environmental regulations and reducing the overhead costs associated with waste treatment and disposal.
Scalability is another major strength of this process, as the reaction does not exhibit the exothermic spikes often seen in vigorous cyclizations, making it safer to scale from grams to kilograms and beyond. The robustness of the catalyst system means that slight variations in mixing or addition rates are less likely to cause batch failures, providing a wider operating window for manufacturing teams. This reliability ensures that production schedules can be met consistently, reducing lead time for high-purity pharmaceutical intermediates and allowing downstream drug manufacturers to plan their synthesis campaigns with greater confidence. The ability to produce diverse analogs using the same core platform chemistry also allows for flexible inventory management, where a single production line can be adapted to make different derivatives based on market demand.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and scope of this silver-catalyzed cyclization technology. These answers are derived directly from the experimental data and claims presented in the patent documentation, providing clarity on the practical aspects of the synthesis. Understanding these details helps stakeholders assess the feasibility of adopting this method for their specific project requirements and ensures alignment between R&D expectations and manufacturing capabilities.
Q: What catalyst system is used for this cyclization reaction?
A: The process utilizes silver nitrate (AgNO3) as the transition metal catalyst, typically at 50 mol%, in conjunction with an organic base like triethylamine.
Q: What are the typical reaction conditions for this synthesis?
A: The reaction proceeds under mild conditions, specifically at room temperature (25°C) in toluene solvent, with a reaction time ranging from 24 to 72 hours depending on the substrate.
Q: What is the scope of substituents compatible with this method?
A: The method tolerates a wide range of functional groups including aryl, heteroaryl, alkyl, ester, cyano, and nitro groups on both the enyne and dicarbonyl components.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 1-Trifluoromethyl-Tetra-Substituted Cyclopentene Derivative Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic value of fluorinated cyclic intermediates in the development of next-generation therapeutics. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that promising laboratory discoveries can be seamlessly transitioned into full-scale manufacturing. We are equipped with rigorous QC labs and adhere to stringent purity specifications to guarantee that every batch of 1-trifluoromethyl-tetra-substituted cyclopentene derivatives meets the highest industry standards. Our commitment to quality and consistency makes us a trusted partner for global pharmaceutical companies seeking to secure their supply chains for critical building blocks.
We invite you to engage with our technical procurement team to discuss how this innovative synthesis route can be tailored to your specific project needs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the economic benefits of switching to this efficient methodology. We encourage potential partners to contact us for specific COA data and route feasibility assessments, allowing us to demonstrate our capability to deliver high-quality fluorinated intermediates that accelerate your drug development timelines while optimizing your overall production costs.