Advanced Catalytic Strategy For Scalable Alpha-Fluorovinyl Sulfide Manufacturing
The development of efficient methodologies for constructing carbon-sulfur bonds in fluorinated scaffolds remains a critical challenge in modern medicinal chemistry, particularly for the production of high-value pharmaceutical intermediates. Patent CN111875523A introduces a groundbreaking palladium-catalyzed protocol that addresses these synthetic hurdles by enabling the direct conversion of trifluoroethyl derivatives into alpha-fluorovinyl sulfide derivatives. This innovation is particularly significant because it bypasses the traditional reliance on unstable and malodorous thiol reagents, instead utilizing odorless and robust diphenyl disulfides as the sulfur source. The process operates under relatively mild thermal conditions, typically between 60-100°C, and employs a cost-effective reductant system involving zinc powder to regenerate the active catalytic species. For R&D directors and process chemists, this represents a substantial leap forward in designing safer, more sustainable routes for complex fluorinated molecules that are essential in drug discovery and agrochemical development.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of vinyl sulfides containing fluorine atoms has been plagued by significant operational and chemical inefficiencies that hinder commercial scalability. Traditional approaches often necessitate the use of free thiols, which are characterized by their extremely unpleasant odors, high volatility, and susceptibility to oxidation, posing severe health and safety risks in industrial settings. Furthermore, achieving selective mono-defluorination of trifluoromethyl groups is notoriously difficult; conventional strong base-mediated methods frequently lead to uncontrolled poly-defluorination or decomposition of the sensitive fluorinated backbone. These legacy processes often require cryogenic temperatures, strictly anhydrous conditions that are expensive to maintain, and complex multi-step sequences to install the sulfur moiety, resulting in poor overall atom economy and excessive waste generation. The inability to tolerate diverse functional groups on the aromatic ring further limits the utility of these older methods, forcing chemists to employ tedious protection and deprotection strategies that drive up manufacturing costs and extend lead times.
The Novel Approach
The methodology disclosed in the patent data offers a transformative solution by leveraging a palladium-catalyzed defluorinative thiolation strategy that is both operationally simple and chemically robust. By employing diphenyl disulfide as a stable sulfur surrogate, the process eliminates the handling issues associated with thiols while maintaining high reactivity through in situ generation of thiolate anions. The reaction proceeds via a streamlined one-pot sequence where oxidative addition of the palladium catalyst to the carbon-bromine bond is followed by a crucial beta-fluorine elimination step, effectively installing the double bond and the sulfur group simultaneously. This approach not only simplifies the workflow by removing the need for intermediate isolation but also demonstrates exceptional substrate universality, accommodating a wide array of electronic environments on both the aryl and sulfur-bearing components. The use of zinc powder as a terminal reductant ensures the continuous turnover of the palladium catalyst, allowing for low catalyst loading and reducing the burden of heavy metal contamination in the final product.

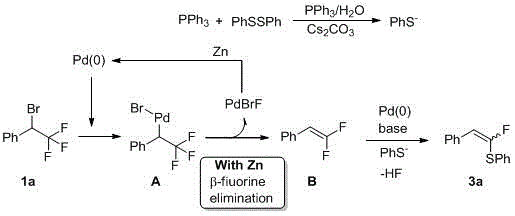
Mechanistic Insights into Pd-Catalyzed Defluorinative Thiolation
A deep understanding of the catalytic cycle is essential for optimizing this process for commercial scale-up of complex pharmaceutical intermediates. The mechanism initiates with the activation of the disulfide bond; in the presence of a base and trace water, diphenyl disulfide reacts with triphenylphosphine to generate the nucleophilic phenylthiolate anion (PhS⁻). Concurrently, the active Pd(0) species undergoes oxidative addition into the carbon-bromine bond of the trifluoroethyl substrate, forming a key organopalladium intermediate. This step is critical as it sets the stage for the subsequent elimination event. Unlike standard cross-couplings, this system is designed to trigger a beta-fluorine elimination from the trifluoromethyl group, which is facilitated by the electron-withdrawing nature of the adjacent palladium center. This elimination releases a molecule of PdBrF and generates a difluoroalkene intermediate, effectively transforming the saturated trifluoroethyl group into a reactive vinyl fluoride motif.
The final stage of the catalytic cycle involves the interception of this difluoroalkene intermediate by the previously generated thiolate anion. This nucleophilic substitution occurs under the promotion of the palladium catalyst or the base present in the medium, leading to the formation of the desired alpha-fluorovinyl sulfide product with high stereoselectivity. Crucially, the PdBrF species generated during the elimination step is reduced back to the active Pd(0) state by the zinc powder, closing the catalytic loop and ensuring sustained reaction efficiency. This mechanistic pathway explains the high yields observed, reaching up to 82% in optimized examples, and highlights why the choice of reductant and base is pivotal. The ability to control the C-F bond activation selectively prevents the formation of fully defluorinated byproducts, ensuring a clean impurity profile that is vital for regulatory compliance in API manufacturing.
How to Synthesize Alpha-Fluorovinyl Sulfide Efficiently
Implementing this synthesis route requires precise control over reaction parameters to maximize yield and minimize side reactions, particularly regarding the stoichiometry of the reductant and the dryness of the solvent. The patent outlines a standardized procedure where the substrate, disulfide, catalyst, ligand, base, and zinc powder are combined in an ultra-dry polar aprotic solvent such as N,N-dimethylformamide. Maintaining an inert nitrogen atmosphere is essential to prevent the oxidation of the phosphine ligand and the palladium catalyst, which could otherwise lead to stalled reactions. While the general protocol is robust, fine-tuning the temperature between 60°C and 100°C allows for accommodation of different substrate reactivities, with 80°C often providing the optimal balance between reaction rate and selectivity. For detailed operational specifics and safety guidelines, please refer to the standardized synthesis steps provided below.
- Combine trifluoroethyl derivative substrate, diphenyl disulfide, palladium acetate catalyst, triphenylphosphine ligand, cesium carbonate base, and zinc powder in ultra-dry DMF solvent.
- Stir the reaction mixture under a nitrogen atmosphere at a temperature range of 60-100°C for approximately 12 hours to facilitate oxidative addition and beta-fluorine elimination.
- Upon completion, quench the mixture into ethyl acetate, wash with saturated brine, dry over anhydrous sodium sulfate, and purify the crude residue via silica gel column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
From a supply chain and procurement perspective, this patented technology offers distinct logistical and economic benefits that directly impact the bottom line of chemical manufacturing operations. The shift from volatile thiols to solid, odorless disulfides drastically reduces the complexity of raw material storage and handling, eliminating the need for specialized scrubbing systems required for malodorous gases. This change not only lowers capital expenditure on safety infrastructure but also accelerates the onboarding of new production lines by simplifying regulatory approvals related to environmental emissions. Furthermore, the one-pot nature of the reaction significantly reduces solvent consumption and energy usage by removing intermediate workup and purification steps, leading to a leaner manufacturing process with a smaller physical footprint. These efficiencies translate into a more resilient supply chain capable of responding rapidly to market demands without the bottlenecks associated with multi-step synthetic sequences.
- Cost Reduction in Manufacturing: The elimination of expensive and hazardous thiol reagents in favor of cheap, commodity-grade diphenyl disulfides results in substantial raw material cost savings. Additionally, the use of zinc powder as a stoichiometric reductant is far more economical than using silanes or other specialized reducing agents often found in similar transformations. The high atom economy of the defluorinative approach means that less starting material is wasted as byproduct, further driving down the cost per kilogram of the final API intermediate. By simplifying the purification process through high-selectivity catalysis, the consumption of silica gel and eluents during chromatography is also minimized, contributing to overall operational expense reduction.
- Enhanced Supply Chain Reliability: Utilizing widely available and stable starting materials mitigates the risk of supply disruptions that are common with specialty fluorinated reagents. The robustness of the catalytic system against moisture and air, relative to other organometallic methods, ensures consistent batch-to-batch quality even in large-scale reactors where perfect inertness is challenging to maintain. This reliability allows for longer campaign runs and reduces the frequency of failed batches, securing a steady flow of critical intermediates for downstream drug synthesis. The broad substrate scope also means that a single manufacturing platform can be adapted to produce a variety of analogues, providing flexibility to pivot production based on changing pipeline priorities without retooling.
- Scalability and Environmental Compliance: The process generates minimal hazardous waste, primarily consisting of zinc salts which are easier to treat and dispose of compared to the complex organic waste streams from traditional thiol chemistry. The absence of foul-smelling emissions aligns with increasingly stringent environmental regulations, reducing the risk of community complaints and regulatory fines. Scalability is further supported by the exothermic profile of the reaction, which is manageable under standard cooling conditions, allowing for safe transition from gram-scale laboratory optimization to ton-scale commercial production. This environmental and safety profile makes the technology highly attractive for green chemistry initiatives and sustainable manufacturing certifications.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and scope of this synthesis technology, derived directly from the experimental data and mechanistic understanding of the patent. These insights are intended to assist technical teams in evaluating the feasibility of adopting this route for their specific project needs. Understanding the nuances of catalyst loading, base selection, and substrate tolerance is key to successful technology transfer.
Q: What is the primary advantage of using diphenyl disulfide over thiols in this synthesis?
A: Diphenyl disulfide is odorless, stable, and commercially inexpensive compared to volatile and malodorous thiols, significantly improving operational safety and reducing environmental hazards in large-scale manufacturing.
Q: How does the catalytic system manage the selective activation of C-F bonds?
A: The system utilizes a palladium catalyst with zinc powder as a reductant to promote selective beta-fluorine elimination after oxidative addition, preventing over-defluorination and ensuring high mono-fluorinated product selectivity.
Q: Is this method suitable for substrates with sensitive functional groups?
A: Yes, the protocol demonstrates excellent functional group tolerance, successfully accommodating electron-withdrawing and electron-donating groups such as halogens, methoxy, and amino groups on the aromatic rings.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Alpha-Fluorovinyl Sulfide Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic value of advanced fluorination technologies in accelerating drug development timelines. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that promising laboratory discoveries can be seamlessly transitioned into reliable industrial supply. We adhere to stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of alpha-fluorovinyl sulfide meets the highest international standards. Our commitment to quality assurance means that we can provide comprehensive documentation and support for regulatory filings, giving our partners confidence in the integrity of their supply chain.
We invite you to collaborate with us to leverage this cutting-edge synthetic methodology for your next project. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can optimize your production costs and reduce lead time for high-purity pharmaceutical intermediates. Let us be your partner in turning complex chemical challenges into commercial successes.