Breakthrough in Arabinoside Impurity Synthesis for Enhanced Pharmaceutical Quality Control
Breakthrough in Arabinoside Impurity Synthesis for Enhanced Pharmaceutical Quality Control
The rigorous demand for high-purity active pharmaceutical ingredients (APIs) in the oncology and antiviral sectors necessitates the availability of well-characterized reference standards and impurities. Patent CN109776639B introduces a transformative synthetic methodology specifically designed for the preparation of critical arabinoside compound impurities, which are essential for the quality control of drugs such as Fludarabine, Clofarabine, and Nelarabine. This technology addresses a longstanding bottleneck in nucleoside chemistry where the selective manipulation of hydroxyl protecting groups has historically resulted in complex mixtures and poor yields. By leveraging a novel deprotection system involving phenylhydrazine, acetic acid, and ammonium bicarbonate, this invention enables the efficient generation of specific di-hydroxy impurities that were previously difficult to isolate. For pharmaceutical manufacturers and quality control laboratories, access to such high-fidelity impurity standards is not merely a regulatory requirement but a cornerstone of patient safety and drug efficacy validation.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
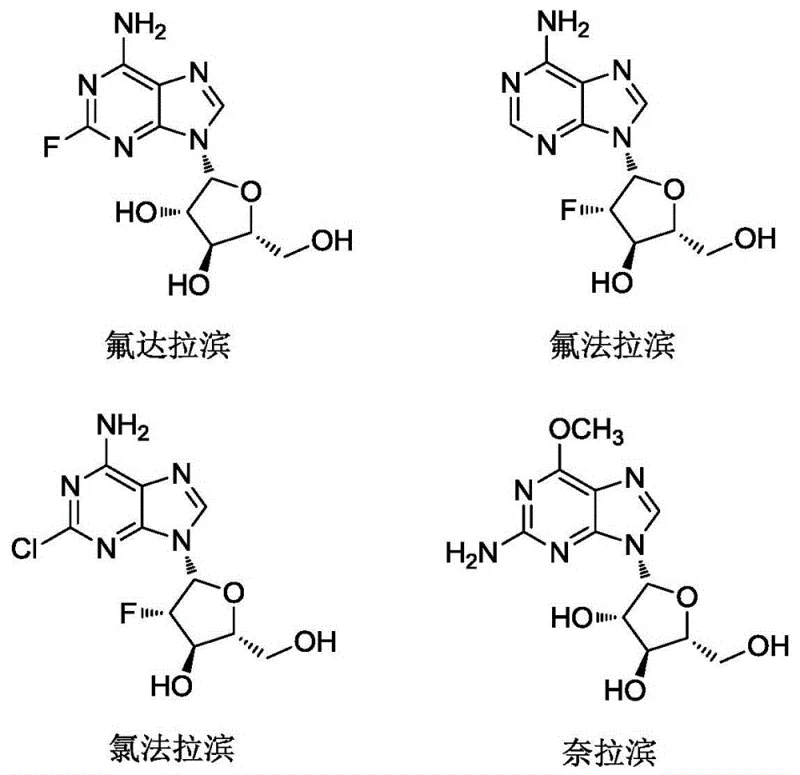
In the traditional synthesis of arabinoside derivatives, the conversion of nucleosides to their arabinose counterparts involves a critical stereochemical inversion at the 2'-position. This process typically requires the protection of multiple hydroxyl groups followed by selective deprotection steps. However, conventional deprotection strategies, such as those described in prior art like US5602246A, suffer from severe selectivity issues. When attempting to remove specific acetyl or benzyl protecting groups, the reaction往往 produces a statistical mixture of mono-, di-, and tri-deprotected species. Specifically, the desired 2',3'-dihydroxy impurity (Formula II) is often formed as a minor byproduct, with yields rarely exceeding 5%. The structural similarity between the target impurity and other isomers, such as the 2'-mono-hydroxy or fully deprotected variants, makes separation via standard crystallization impossible. Consequently, manufacturers are forced to rely on resource-intensive preparative liquid chromatography (Prep-HPLC), which drastically increases production costs, limits scalability, and introduces significant delays in the supply chain for reference materials.
The Novel Approach
The methodology disclosed in CN109776639B represents a paradigm shift by fundamentally altering the chemoselectivity of the deprotection reaction. Instead of relying on basic hydrolysis or standard hydrogenolysis which lack discrimination between the 2', 3', and 5' positions, this invention employs a buffered nucleophilic system. The use of phenylhydrazine in conjunction with acetic acid and ammonium bicarbonate creates a unique chemical environment that favors the simultaneous removal of protecting groups at the 2' and 3' positions while preserving the protection at the 5' position. This results in the target impurity (Formula II) becoming the major product of the reaction, with crude purities exceeding 75%. This dramatic improvement in selectivity eliminates the need for complex chromatographic purification. Instead, the target compound can be isolated through straightforward filtration and recrystallization techniques, achieving final purities greater than 95% with yields around 50%. This approach transforms the synthesis of these critical impurities from a laboratory curiosity into a commercially viable manufacturing process.
Mechanistic Insights into Phenylhydrazine-Mediated Selective Deprotection
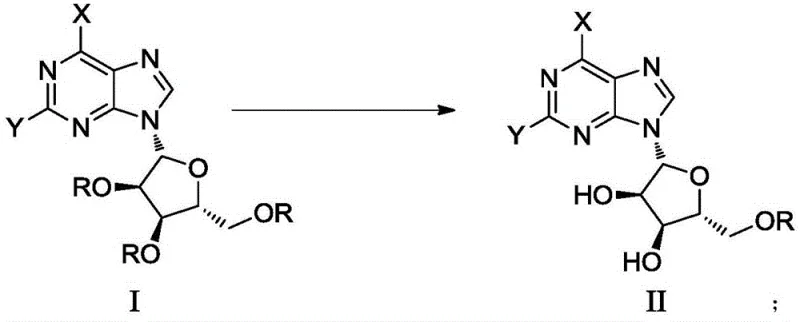
The success of this synthetic route lies in the precise interplay between the nucleophilicity of phenylhydrazine and the buffering capacity of the acetic acid/ammonium bicarbonate system. Phenylhydrazine acts as a potent nucleophile capable of attacking the ester or ether linkages of the protecting groups (such as acetyl or benzyl). However, without pH control, this reaction would proceed indiscriminately, leading to full deprotection. The inclusion of acetic acid and ammonium bicarbonate establishes a buffered medium that modulates the reactivity of the phenylhydrazine. This buffering effect is crucial for differentiating between the steric and electronic environments of the 2', 3', and 5' hydroxyl groups. The 5'-position, being primary and less sterically hindered, might typically be more reactive, but the specific reagent ratios (Substrate:Phenylhydrazine:Acetic Acid:Ammonium Bicarbonate at 1:3-5:1-2:5-10) appear to kinetically favor the cleavage at the secondary 2' and 3' positions. This suggests a mechanism where the reagents coordinate with the sugar ring in a specific conformation that exposes the 2' and 3' protecting groups to nucleophilic attack while shielding the 5' position, or where the thermodynamic stability of the 5'-protected intermediate is maximized under these specific conditions.
Furthermore, the control of side reactions is a critical aspect of this mechanism. In conventional processes, the formation of Formula III (2'-mono-deprotected) and Formula IV (fully deprotected) compounds is prevalent, complicating the purification landscape. The new method effectively suppresses the formation of these unwanted byproducts. By maintaining the reaction temperature within a mild range of 10°C to 25°C, the kinetic energy of the system is kept low enough to prevent non-selective thermal degradation or over-reaction, yet high enough to allow the catalyzed deprotection to proceed. The solvent system, utilizing pyridine or pyridine blends with DMF or DCM, further stabilizes the transition states involved in the deprotection. Pyridine, acting as both a solvent and a weak base, likely assists in the proton transfer steps necessary for the cleavage of the protecting groups. This multi-factorial control over reagent stoichiometry, temperature, and solvent environment ensures that the impurity profile of the crude reaction mixture is heavily skewed towards the desired target, simplifying downstream processing significantly.
How to Synthesize Arabinoside Impurities Efficiently
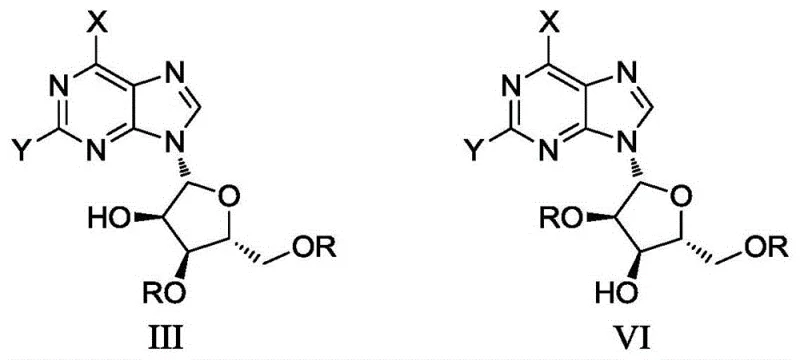
The implementation of this synthesis protocol requires careful attention to reagent quality and stoichiometric precision to replicate the high selectivity reported in the patent. The process begins with the dissolution of the tri-protected arabinoside starting material in a suitable solvent system, typically involving pyridine to ensure complete solubility and participation in the reaction mechanism. The addition of the deprotection cocktail must be managed to maintain the specific molar ratios identified as optimal, as deviations can lead to a resurgence of the traditional impurity profile dominated by Formula III. Following the reaction period, which typically spans 18 to 24 hours, the workup procedure is notably simple compared to chromatographic methods. The use of toluene for dispersion and precipitation allows for the bulk removal of soluble byproducts and excess reagents. Subsequent recrystallization steps using water/ethanol or water/acetone mixtures polish the crude material to reference standard grade purity. For detailed operational parameters and safety considerations regarding the handling of phenylhydrazine, please refer to the standardized synthesis guide below.
- Prepare the reaction mixture by dissolving the 2',3',5'-tri-protected arabinoside substrate in a solvent system containing pyridine or pyridine/DMF blends.
- Add the specific deprotection reagents: phenylhydrazine, acetic acid, and ammonium bicarbonate in a controlled molar ratio to ensure selective 2',3'-deprotection.
- Maintain mild reaction temperatures between 10°C and 25°C, followed by workup involving toluene dispersion and recrystallization to achieve >95% purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors in the pharmaceutical sector, the adoption of this synthetic method offers substantial strategic benefits beyond mere technical feasibility. The primary advantage lies in the drastic simplification of the purification workflow. By shifting the isolation method from preparative HPLC to crystallization, the cost of goods sold (COGS) for these high-value impurities is significantly reduced. Preparative chromatography is not only capital intensive, requiring expensive columns and solvents, but also operationally slow, creating bottlenecks in production throughput. Eliminating this step allows for larger batch sizes and faster turnaround times, ensuring a more reliable supply of critical reference materials. Furthermore, the use of commodity chemicals like phenylhydrazine, acetic acid, and ammonium bicarbonate reduces dependency on exotic or proprietary catalysts, mitigating supply chain risks associated with single-source vendors. This robustness in raw material sourcing enhances the overall resilience of the supply chain against market fluctuations.
- Cost Reduction in Manufacturing: The economic impact of this technology is driven by the elimination of chromatographic purification. Traditional methods yielding less than 5% purity require massive amounts of starting material and solvent to isolate milligram quantities of the target impurity. In contrast, the new method achieves yields around 50% with high crude purity. This ten-fold increase in effective yield translates directly to lower raw material consumption and reduced waste disposal costs. Additionally, the avoidance of specialized liquid-mass separation equipment lowers the barrier to entry for production, allowing for manufacturing in standard multipurpose reactors rather than dedicated purification suites. The reduction in solvent usage, particularly the avoidance of large volumes of HPLC-grade solvents, further contributes to a leaner cost structure and improved environmental footprint.
- Enhanced Supply Chain Reliability: Supply continuity for reference standards is often jeopardized by the complexity of their synthesis. The simplified workflow of this new method, characterized by mild reaction conditions (10-25°C) and ambient pressure operations, reduces the risk of batch failures due to equipment malfunction or parameter deviation. The robustness of the reaction against minor variations in conditions ensures consistent output quality, which is vital for maintaining long-term contracts with pharmaceutical clients. Moreover, the ability to source reagents from multiple global suppliers prevents disruptions caused by regional shortages. This reliability is crucial for API manufacturers who depend on timely delivery of impurity standards to validate their own production batches and meet regulatory filing deadlines without delay.
- Scalability and Environmental Compliance: From a scale-up perspective, the transition from lab to pilot to commercial scale is streamlined by the absence of chromatography. Crystallization is a unit operation that scales linearly and predictably, unlike chromatography which often faces mass transfer limitations at larger scales. This facilitates the rapid expansion of production capacity to meet growing market demand for arabinoside drugs. Environmentally, the process aligns with green chemistry principles by reducing solvent intensity and waste generation. The ability to recycle mother liquors from the crystallization steps further minimizes the environmental burden. Compliance with increasingly stringent environmental regulations is easier to achieve with a process that avoids the massive solvent waste streams associated with preparative HPLC, thereby reducing the liability and operational costs related to waste treatment and disposal.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the synthesis and application of arabinoside impurities. These answers are derived from the specific technical disclosures and experimental data found within the patent literature, providing a factual basis for decision-making. Understanding the nuances of this selective deprotection chemistry is essential for stakeholders evaluating the feasibility of integrating these standards into their quality control frameworks. The clarity provided here aims to bridge the gap between complex organic synthesis and practical supply chain management.
Q: Why is synthesizing arabinoside impurities technically challenging?
A: Traditional deprotection methods lack selectivity, often producing mixtures of 2'-mono-deprotected, 2',3'-di-deprotected, and fully deprotected species. Separating these structurally similar isomers typically requires expensive preparative HPLC with very low yields.
Q: What is the key advantage of the phenylhydrazine-based method?
A: This method utilizes a unique reagent combination (phenylhydrazine/acetic acid/ammonium bicarbonate) that shifts the reaction selectivity dramatically, making the desired 2',3'-dihydroxy impurity the major product rather than a minor byproduct.
Q: Can this process be scaled for commercial reference standard production?
A: Yes, the process operates under mild conditions (10-25°C) and uses common solvents like pyridine and toluene. The ability to purify via simple recrystallization instead of chromatography makes it highly scalable for industrial supply.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Arabinoside Impurity Supplier
At NINGBO INNO PHARMCHEM, we recognize that the integrity of your pharmaceutical products depends on the quality of your reference standards. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that even complex molecules like arabinoside impurities are manufactured with the highest degree of precision. We adhere to stringent purity specifications and utilize rigorous QC labs to verify the identity and purity of every batch, guaranteeing that our materials meet the demanding requirements of global regulatory agencies. Our commitment to excellence extends beyond simple manufacturing; we act as a strategic partner in your drug development lifecycle, providing the critical materials needed to ensure patient safety and product efficacy.
We invite you to leverage our expertise to optimize your supply chain for nucleoside analogs. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific impurity profiling needs. We are prepared to provide specific COA data and route feasibility assessments to demonstrate how our advanced synthetic capabilities can enhance your operational efficiency. Let us help you secure a stable, high-quality supply of arabinoside intermediates and impurities, enabling you to focus on what matters most: delivering life-saving therapies to patients worldwide.