Advanced ZP-1609 Manufacturing: Streamlined Synthesis for Commercial Scale-Up
Advanced ZP-1609 Manufacturing: Streamlined Synthesis for Commercial Scale-Up
The pharmaceutical landscape for cardiovascular therapeutics is constantly evolving, driven by the need for more efficient manufacturing processes that ensure high purity and supply stability. A significant advancement in this domain is detailed in patent CN112142823B, which discloses a novel synthesis method for ZP-1609, also known as Danegaptide. This small-molecule modified dipeptide serves as a critical GAP junction modifier with potential antiarrhythmic efficacy, currently undergoing clinical evaluation for treating ST-elevation myocardial infarction (STEMI) and preventing atrial fibrillation. The disclosed technology represents a paradigm shift from traditional multi-step protection strategies to a streamlined approach that leverages the inherent reactivity of specific precursors. By optimizing the synthetic pathway, this method addresses long-standing challenges in peptide intermediate production, such as low overall yields and complex purification protocols. For stakeholders in the fine chemical industry, understanding this technological leap is essential for evaluating future supply chain resilience and cost structures associated with high-value cardiac drug intermediates.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of complex dipeptides like ZP-1609 has been plagued by inefficient protection-deprotection sequences that inflate production costs and extend lead times. Conventional routes, as illustrated in prior art literature such as J. Med. Chem. 2009, typically commence with fully protected starting materials, specifically (2S,4R)-1-tert-butyloxycarbonyl-4-aminopyrrolidine-2-carboxylic acid methyl ester. This necessitates an initial amidation step followed by a dedicated deprotection of the N-terminal amino group before the glycine moiety can be linked. Furthermore, the use of acyl chlorides in traditional amidation reactions introduces harsh reaction conditions that often generate significant byproducts, complicating downstream purification. The requirement for multiple discrete deprotection steps, often involving different reagents for the N-terminus and C-terminus, not only reduces the overall atom economy but also increases the risk of racemization and impurity formation. These cumulative inefficiencies create a bottleneck for manufacturers aiming to produce high-purity pharmaceutical intermediates at a commercial scale.
The Novel Approach
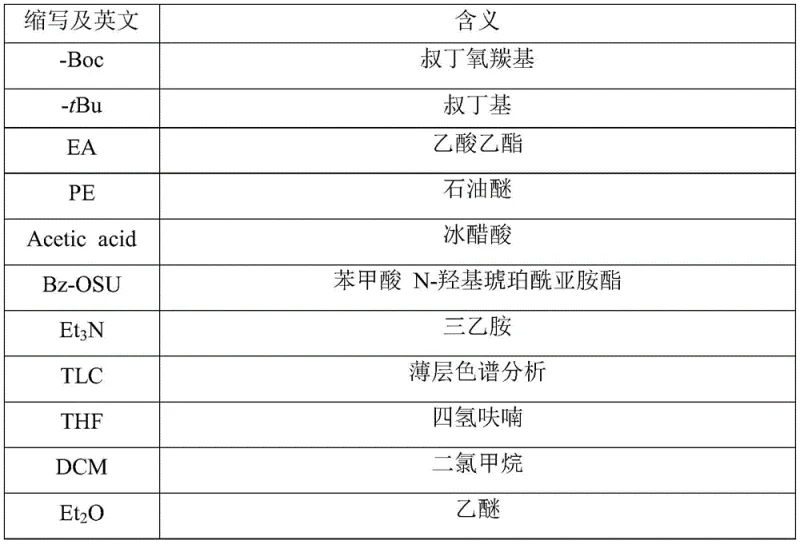
In stark contrast to the cumbersome legacy pathways, the methodology described in patent CN112142823B introduces a highly efficient strategy that fundamentally simplifies the molecular construction of ZP-1609. The innovation lies in the strategic selection of 4-amino-L-proline tert-butyl ester as the starting material, which possesses a free amino group ready for immediate selective reaction, thereby bypassing the need for initial N-end protection entirely. This approach allows for the direct formation of the benzamide bond using benzoic acid N-hydroxysuccinimide ester under mild conditions, significantly mitigating the formation of side products associated with acyl chlorides. Subsequently, the glycine fragment is introduced via a robust DCC/HOBT mediated coupling, ensuring high fidelity in peptide bond formation. Perhaps most critically, the final step exploits the acid lability of both the Boc group on glycine and the tert-butyl ester on proline, enabling a simultaneous one-pot hydrolysis and acidification. This consolidation of steps drastically reduces solvent usage, labor intensity, and processing time, offering a compelling value proposition for industrial adoption.
Mechanistic Insights into Selective Amidation and Acidolytic Deprotection
The core chemical elegance of this synthesis lies in the precise control of chemoselectivity during the initial amidation phase. By utilizing benzoic acid N-hydroxysuccinimide ester (Bz-OSU) as the acylating agent, the reaction proceeds through a highly reactive active ester intermediate that selectively targets the primary amine of the proline derivative without affecting the tert-butyl ester moiety. This selectivity is paramount, as it preserves the C-terminal protecting group for the final global deprotection step. The reaction environment, typically maintained in a biphasic or mixed solvent system involving water and tetrahydrofuran with triethylamine as a base, ensures that the nucleophilic attack occurs efficiently while minimizing hydrolysis of the active ester. Following this, the introduction of the glycine unit via DCC and HOBT facilitates the formation of the peptide bond through an O-acylisourea intermediate, which is stabilized by HOBT to prevent racemization—a common pitfall in peptide synthesis. The resulting intermediate retains orthogonal protecting groups that are strategically aligned for the final transformation.
The final mechanistic breakthrough involves the concurrent removal of protecting groups under acidic conditions. Both the tert-butoxycarbonyl (Boc) group on the glycine nitrogen and the tert-butyl (tBu) ester on the proline carboxyl terminus share a common vulnerability to strong acids like hydrochloric acid. In the patented process, treating the fully assembled dipeptide intermediate with 4N HCl triggers a cascade of cleavage reactions where the carbamate and ester bonds are hydrolyzed simultaneously. This generates the free amine and free carboxylic acid functionalities required for the final zwitterionic structure of ZP-1609, while also converting the amine into its hydrochloride salt form in situ. This one-pot acidolysis eliminates the need for intermediate isolation and separate deprotection reagents, thereby streamlining the workflow and enhancing the purity profile of the final API intermediate by reducing the number of unit operations where impurities could be introduced.
How to Synthesize ZP-1609 Efficiently
The implementation of this synthesis route requires careful attention to stoichiometry and reaction monitoring to maximize the benefits of the streamlined design. The process begins with the selective acylation of the proline derivative, followed by peptide coupling, and concludes with the global deprotection. Each stage is optimized for high conversion and ease of purification, often relying on simple recrystallization techniques rather than complex chromatography. For process chemists looking to adopt this methodology, the following guide outlines the critical operational parameters derived from the patent examples to ensure successful replication and scale-up.
- Selectively react 4-amino-L-proline tert-butyl ester with benzoic acid N-hydroxysuccinimide ester to form the benzamide intermediate.
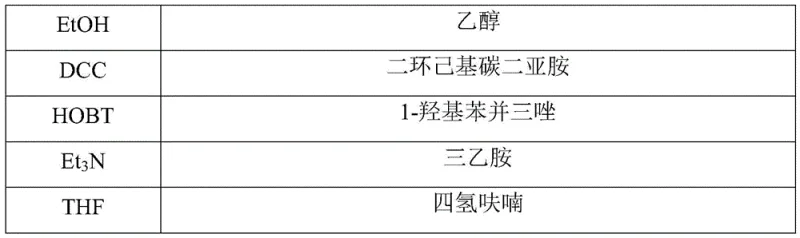
- Couple the intermediate with Boc-Gly-OH using DCC and HOBT in THF to establish the peptide bond.
- Perform a one-step hydrolysis with hydrochloric acid to simultaneously remove Boc and tBu protecting groups and acidify the final product.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, the adoption of this novel synthesis route offers substantial strategic advantages for procurement managers and supply chain directors overseeing the sourcing of cardiovascular drug intermediates. The reduction in synthetic steps directly correlates to a decrease in raw material consumption, solvent waste, and energy usage, which collectively drive down the cost of goods sold (COGS). By eliminating the need for harsh acyl chlorides and multiple protection/deprotection cycles, the process becomes inherently safer and more environmentally compliant, reducing the regulatory burden associated with hazardous waste disposal. Furthermore, the simplified workflow enhances manufacturing throughput, allowing producers to respond more agilely to market demand fluctuations without compromising on quality standards. These factors combine to create a more resilient supply chain capable of delivering high-purity materials with greater reliability.
- Cost Reduction in Manufacturing: The elimination of discrete protection and deprotection steps significantly lowers the operational expenditure associated with ZP-1609 production. By removing the necessity for N-terminal protection on the starting proline material and combining the final deprotection with acidification, the process reduces the consumption of expensive protecting group reagents and the solvents required for intermediate isolations. Additionally, the shift from acyl chlorides to active esters minimizes the generation of salt byproducts, simplifying the workup procedure and reducing the load on wastewater treatment systems. This leaner manufacturing footprint translates into tangible cost savings that can be passed down the supply chain, making the final API more economically viable for pharmaceutical developers.
- Enhanced Supply Chain Reliability: The reliance on readily available and stable starting materials, such as 4-amino-L-proline tert-butyl ester and Boc-Gly-OH, mitigates the risk of supply disruptions often caused by specialized or hazardous reagents. The robustness of the DCC/HOBT coupling chemistry ensures consistent reaction outcomes, reducing the likelihood of batch failures that can delay delivery schedules. Moreover, the ability to purify intermediates and the final product through straightforward recrystallization, rather than resource-intensive chromatography, accelerates the production cycle time. This operational efficiency ensures a steady flow of materials, providing downstream pharmaceutical manufacturers with the confidence needed to plan their own clinical and commercial production timelines effectively.
- Scalability and Environmental Compliance: The mild reaction conditions employed throughout this synthesis, particularly the avoidance of cryogenic temperatures and highly corrosive reagents, make the process exceptionally well-suited for large-scale industrial production. The one-pot final deprotection step not only saves time but also significantly reduces the volume of chemical waste generated per kilogram of product, aligning with modern green chemistry principles. This environmental advantage simplifies compliance with increasingly stringent global regulations regarding pharmaceutical manufacturing emissions. Consequently, facilities can scale up production from pilot batches to multi-ton annual capacities with minimal need for specialized infrastructure upgrades, ensuring long-term sustainability and operational flexibility.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production of ZP-1609, based on the detailed specifications and experimental data provided in the patent literature. These insights are intended to clarify the practical implications of the new synthesis method for industry professionals evaluating this technology for potential integration into their supply networks.
Q: How does the new ZP-1609 synthesis route improve upon conventional methods?
A: The novel route eliminates the need for N-terminal protection on the proline starting material and combines the final deprotection and acidification into a single step, reducing the total number of synthetic operations and improving overall yield.
Q: What are the key reagents used in the optimized ZP-1609 manufacturing process?
A: The process utilizes 4-amino-L-proline tert-butyl ester, benzoic acid N-hydroxysuccinimide ester (Bz-OSU), Boc-Gly-OH, DCC, HOBT, and hydrochloric acid, avoiding harsh acyl chloride reagents.
Q: Is the ZP-1609 synthesis method scalable for industrial production?
A: Yes, the method employs mild reaction conditions, liquid-phase amide condensation, and simple recrystallization purification, making it highly suitable for commercial scale-up and consistent quality control.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable ZP-1609 Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthesis routes in the development of life-saving cardiovascular therapies. Our team of expert process chemists has thoroughly analyzed the technological advancements presented in patent CN112142823B and is fully equipped to translate this innovative methodology into commercial reality. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to industrial reactor is seamless and robust. Our state-of-the-art facilities are designed to handle complex peptide intermediates with stringent purity specifications, supported by rigorous QC labs that employ advanced analytical techniques to guarantee batch-to-batch consistency and compliance with international pharmacopoeia standards.
We invite pharmaceutical partners and procurement leaders to engage with us for a Customized Cost-Saving Analysis tailored to your specific ZP-1609 requirements. By leveraging our optimized manufacturing capabilities, we can help you reduce lead times and secure a stable supply of high-quality intermediates essential for your clinical and commercial programs. Please contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate how our commitment to technical excellence can drive value for your organization.