Scalable Synthesis of 2-(2,5-Difluorophenyl)pyrrolidine Hydrochloride for Oncology APIs
Scalable Synthesis of 2-(2,5-Difluorophenyl)pyrrolidine Hydrochloride for Oncology APIs
The pharmaceutical industry continuously seeks robust and scalable pathways for complex oncology intermediates, particularly those serving tyrosine kinase inhibitors like larotrectinib. Patent CN112250611B introduces a transformative synthetic methodology for producing 2-(2,5-difluorophenyl)pyrrolidine hydrochloride, a critical chiral precursor. This innovation addresses long-standing challenges in process safety and raw material availability by shifting away from hazardous organometallic chemistries toward a more benign Friedel-Crafts based strategy. For R&D directors and supply chain leaders, this patent represents a significant opportunity to optimize the manufacturing of high-purity pharmaceutical intermediates while mitigating the risks associated with traditional Grignard-based protocols. The following analysis details the technical merits and commercial viability of this novel four-step sequence.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
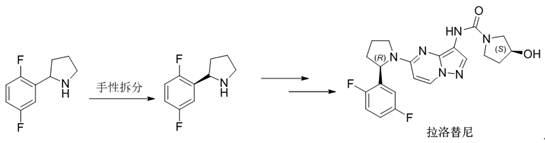
Historically, the synthesis of 2-(2,5-difluorophenyl)pyrrolidine has relied heavily on organometallic coupling reactions that pose significant barriers to industrial implementation. As illustrated in prior art such as Chinese Patent CN201810066842.5, traditional routes often involve the generation of Grignard reagents from 2,5-difluorobromobenzene, which necessitates strictly anhydrous and oxygen-free environments to prevent catastrophic failure or fire hazards. Furthermore, alternative pathways described in US2016168156A1 utilize expensive and less accessible starting materials like N-Boc-2-pyrrolidone derivatives, which drive up the cost of goods sold (COGS) and complicate procurement logistics. These conventional methods not only demand specialized reactor capabilities for handling pyrophoric substances but also frequently suffer from inconsistent yields and difficult impurity profiles due to the sensitivity of the metal-halogen exchange steps.
The Novel Approach
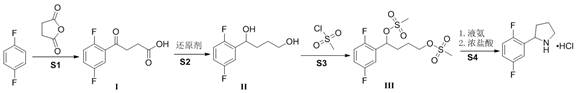
In stark contrast, the methodology disclosed in CN112250611B leverages a Friedel-Crafts acylation between p-difluorobenzene and succinic anhydride as the foundational step, utilizing commodity chemicals that are abundant in the global fine chemical market. This strategic shift eliminates the need for cryogenic temperatures and moisture-sensitive reagents, allowing the reaction to proceed under mild thermal conditions (35-38°C) with aluminum trichloride as a catalyst. The subsequent transformation involves a streamlined reduction and cyclization sequence that avoids the use of transition metals or complex protecting group strategies found in older patents. By adopting this route, manufacturers can achieve a total process yield exceeding 60% with intermediate purities consistently above 98%, thereby offering a reliable pharmaceutical intermediate supplier solution that balances technical excellence with economic efficiency.
Mechanistic Insights into Friedel-Crafts Acylation and Reductive Cyclization
The core of this innovative process lies in the initial electrophilic aromatic substitution, where p-difluorobenzene reacts with succinic anhydride in the presence of anhydrous aluminum trichloride to form the keto-acid Intermediate I. This step is critical for establishing the carbon skeleton, and the choice of dichloromethane as a solvent ensures optimal solubility and heat dissipation during the exothermic acylation. Following isolation, Intermediate I undergoes a robust reduction using borane-tetrahydrofuran complexes or sodium borohydride systems to yield the corresponding diol, Intermediate II. The mechanistic precision here allows for the complete reduction of the ketone functionality without affecting the aromatic fluorine substituents, a common pitfall in less selective reduction protocols. This high chemoselectivity is paramount for maintaining the structural integrity required for downstream chiral resolution into the active (2R)-enantiomer used in larotrectinib synthesis.
The final stages involve the activation of the diol via mesylation with methanesulfonyl chloride, followed by a nucleophilic substitution cyclization using liquid ammonia. This intramolecular ring-closing step is particularly elegant, as it simultaneously constructs the pyrrolidine ring and introduces the nitrogen atom in a single operation under moderate pressure (0.4-0.5 MPa). The use of liquid ammonia serves a dual purpose as both the nucleophile and the solvent, driving the equilibrium towards the desired cyclic amine while minimizing the formation of oligomeric byproducts. Subsequent salification with hydrochloric acid precipitates the final product as a stable hydrochloride salt, ensuring excellent storage stability and ease of handling. This mechanism effectively controls the impurity profile by avoiding radical pathways or high-energy intermediates that typically generate hard-to-remove trace contaminants.
How to Synthesize 2-(2,5-Difluorophenyl)pyrrolidine Hydrochloride Efficiently
Implementing this synthesis requires careful attention to the stoichiometry of the Lewis acid catalyst and the control of exotherms during the acylation phase. The patent outlines a clear progression from acylation to reduction, activation, and finally cyclization, with specific workup procedures designed to maximize recovery at each stage. Operators should note the importance of the recrystallization steps using toluene and methanol, which are essential for achieving the reported purity levels of ≥98% for all intermediates. The detailed standardized synthesis steps below provide a roadmap for replicating this high-efficiency process in a pilot or production setting.
- Perform Friedel-Crafts reaction between p-difluorobenzene and succinic anhydride using AlCl3 catalyst to obtain Intermediate I.
- Reduce Intermediate I using borane-tetrahydrofuran or sodium borohydride systems to generate the diol Intermediate II.
- React Intermediate II with methanesulfonyl chloride in the presence of a base to form the bis-mesylate Intermediate III.
- Cyclize Intermediate III with liquid ammonia under pressure (0.4-0.5 MPa) followed by salification with HCl to yield the final product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this patented route offers substantial strategic benefits beyond mere technical feasibility. The primary advantage lies in the drastic simplification of the raw material basket; by replacing specialized, low-volume reagents with bulk commodities like p-difluorobenzene and succinic anhydride, companies can significantly reduce exposure to supply volatility and price fluctuations. This shift not only lowers the direct material costs but also simplifies vendor qualification processes, as these starting materials are produced by multiple global suppliers with established quality standards. Furthermore, the elimination of pyrophoric Grignard reagents removes the need for specialized containment infrastructure and hazardous waste disposal protocols, leading to indirect cost savings in facility maintenance and regulatory compliance.
- Cost Reduction in Manufacturing: The economic impact of this process is driven by the avoidance of expensive protecting groups and the use of catalytic rather than stoichiometric metal reagents in key steps. By removing the requirement for ultra-low temperature cooling systems needed for organolithium or Grignard reactions, energy consumption is drastically lowered, contributing to a leaner manufacturing cost structure. Additionally, the high yield of each individual step minimizes the loss of valuable fluorinated intermediates, ensuring that the overall cost per kilogram of the final API intermediate is optimized for commercial competitiveness.
- Enhanced Supply Chain Reliability: Reliance on commodity chemicals ensures that production schedules are not held hostage by the lead times of niche custom synthesis providers. The robustness of the reaction conditions means that batch-to-batch variability is minimized, reducing the risk of production delays caused by failed runs or out-of-specification results. This reliability is crucial for maintaining continuous supply to downstream API manufacturers, particularly in the oncology sector where demand is inelastic and timely delivery is critical for patient access.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, utilizing standard reactor types and avoiding exotic conditions that are difficult to translate from lab to plant. The waste stream is primarily composed of aqueous salts and organic solvents that can be readily treated or recycled, aligning with modern green chemistry principles and reducing the environmental footprint of the manufacturing site. This compliance with environmental standards facilitates smoother regulatory approvals and enhances the corporate sustainability profile of the manufacturing entity.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis route. These answers are derived directly from the experimental data and beneficial effects reported in the patent documentation, providing clarity on safety, purity, and scalability concerns.
Q: What are the key safety advantages of this new synthesis route compared to Grignard methods?
A: The patented route eliminates the need for pyrophoric Grignard reagents and strictly anhydrous/oxygen-free conditions required in conventional methods. By utilizing Friedel-Crafts acylation and liquid ammonia cyclization at moderate pressures (0.4-0.5 MPa), the process significantly reduces operational hazards and simplifies equipment requirements for industrial scale-up.
Q: How does this method improve the purity profile of the pharmaceutical intermediate?
A: The stepwise approach allows for rigorous purification at each stage, with reported intermediate purities consistently exceeding 98%. The use of recrystallization (toluene/methanol) and distillation steps effectively removes side products, ensuring the final 2-(2,5-difluorophenyl)pyrrolidine hydrochloride meets stringent specifications for oncology drug synthesis.
Q: Are the raw materials for this process readily available for large-scale procurement?
A: Yes, the synthesis relies on commodity chemicals such as p-difluorobenzene, succinic anhydride, and methanesulfonyl chloride. Unlike prior art routes that require specialized and expensive precursors like N-Boc-2-pyrrolidone derivatives, this method utilizes widely sourced bulk chemicals, enhancing supply chain stability and reducing raw material costs.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 2-(2,5-Difluorophenyl)pyrrolidine Hydrochloride Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of securing a stable supply of high-quality oncology intermediates. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that we can meet the rigorous demands of global pharmaceutical partners. We are committed to delivering products with stringent purity specifications, supported by our rigorous QC labs that employ advanced analytical techniques to verify every batch against the highest industry standards. Our capability to adapt and optimize synthetic routes like the one described in CN112250611B allows us to offer a secure and efficient supply chain solution for complex pharmaceutical intermediates.
We invite you to collaborate with us to explore how this advanced synthesis method can benefit your specific project requirements. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your volume needs. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing expertise can drive value and efficiency in your supply chain.