Advanced Synthesis of Nilotinib Intermediates: A Breakthrough in Purity and Scalability
The pharmaceutical landscape for chronic myelogenous leukemia (CML) treatment has been significantly shaped by tyrosine kinase inhibitors, with Nilotinib (marketed as Tasigna) standing out as a critical second-generation therapy. The efficient and pure synthesis of its key intermediates remains a paramount challenge for generic manufacturers and CDMOs aiming to secure reliable supply chains. Patent CN107235910B, published in April 2020, discloses a transformative synthesis method for the pivotal intermediate known as nilamide or Compound IV (3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)aniline). This intellectual property addresses long-standing issues regarding impurity control and yield optimization that have plagued earlier synthetic routes. By introducing a strategic amino protection-deprotection sequence, the invention achieves a chemical purity exceeding 99.5%, drastically reducing the burden on downstream purification processes. For R&D directors and procurement specialists, this represents a vital opportunity to enhance the robustness of API manufacturing. The following analysis delves into the technical nuances of this patented route, highlighting its potential to redefine cost structures and supply reliability for high-purity pharmaceutical intermediates.
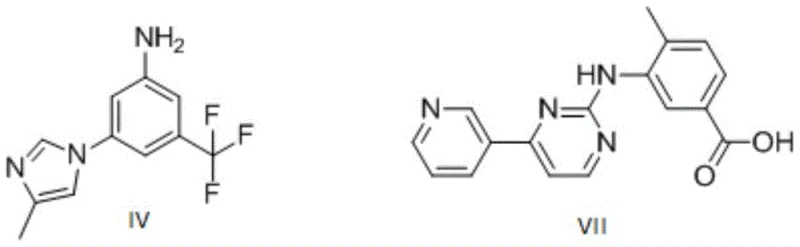
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the preparation of Compound IV has been fraught with significant technical and economic hurdles that hinder efficient commercialization. Early approaches, such as those disclosed in WO2004/005281, relied on a multi-step sequence involving substitution, hydrolysis, and a Curtius rearrangement. While chemically feasible, this route necessitates the use of diphenyl phosphoryl azide (DPPA), a reagent that is not only prohibitively expensive but also poses severe safety risks due to its toxicity and potential explosiveness. Furthermore, these legacy methods often require column chromatography for purification, a unit operation that is notoriously difficult to scale and economically unviable for multi-kilogram or ton-scale production. Alternative routes reported in WO2006/135619 attempted to shorten the step count by using dinitro or bromo-nitro starting materials; however, these suffered from abysmal yields ranging from 21.1% to 35%, rendering them commercially unattractive. Perhaps most critically, direct coupling methods described in WO2006/135640, which react 3-fluoro-5-trifluoromethylaniline directly with imidazole, result in unacceptable impurity profiles. The naked amino group participates in unwanted self-coupling reactions to generate byproduct VIII, and poor regioselectivity leads to approximately 15% of the 5-position isomer impurity (Compound IX). These impurities are difficult to remove without extensive chromatography, compromising both yield and final drug safety.
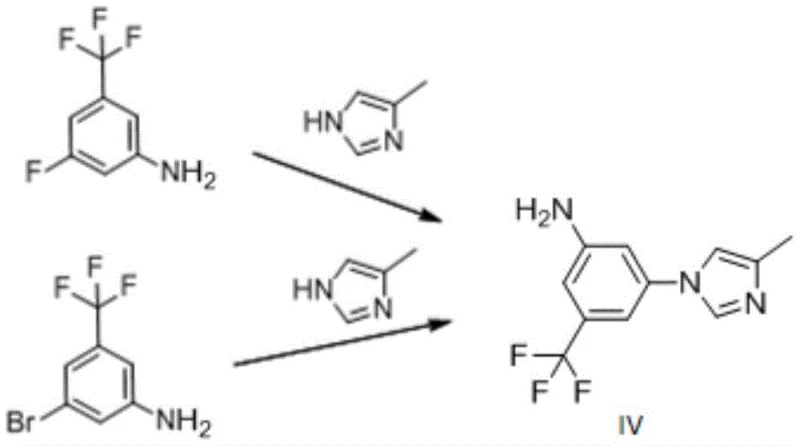
The Novel Approach
In stark contrast to these flawed precedents, the method disclosed in CN107235910B introduces a clever protective group strategy that fundamentally alters the reaction trajectory. The core innovation lies in the initial N-benzylation of the aniline starting material (Compound I) to form Compound II. By masking the nucleophilic amino group with a benzyl moiety, the synthesis effectively prevents the self-coupling side reactions that plague direct methods. Subsequently, the protected intermediate undergoes a copper-catalyzed coupling with 4-methyl-1H-imidazole to form Compound III. This step benefits from the steric and electronic modulation provided by the protecting group, which enhances regioselectivity and suppresses the formation of the troublesome isomer IX. The final step involves a clean debenzylation, achievable through hydrogenation or acidic hydrolysis, to reveal the target aniline. This three-step sequence avoids hazardous reagents like DPPA and eliminates the need for column chromatography, relying instead on crystallization for purification. The result is a process that is not only chemically elegant but also industrially pragmatic, offering a clear pathway to high-purity material suitable for GMP manufacturing environments.
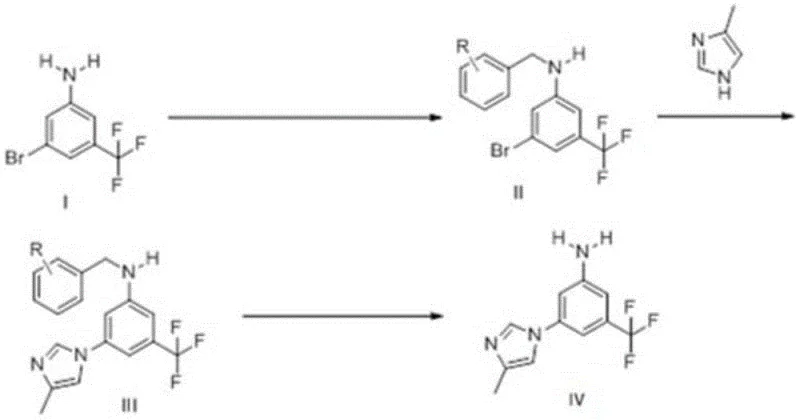
Mechanistic Insights into Copper-Catalyzed Coupling and Impurity Control
The heart of this synthetic advancement is the copper-catalyzed C-N bond formation, likely proceeding via a Ullmann-type or Chan-Lam-type mechanism depending on the specific oxidation state of the copper source used. In the presence of ligands such as 8-hydroxyquinoline or 1,2-cyclohexanediamine, the copper catalyst facilitates the oxidative addition into the carbon-halogen bond of the protected aryl halide (Compound II). The presence of the N-benzyl group is mechanistically crucial; it reduces the electron density on the nitrogen atom of the aniline ring, thereby diminishing its competitiveness as a nucleophile against the imidazole nitrogen. This electronic deactivation is key to preventing the formation of the symmetric self-coupling byproduct (Compound VIII), where two aniline molecules would otherwise link together. Furthermore, the steric bulk of the benzyl group may influence the transition state geometry during the coupling with the imidazole ring, favoring attack at the N1 position of the imidazole over the N3 position, which corresponds to the formation of the desired 4-methyl-1H-imidazol-1-yl isomer rather than the 5-methyl isomer. The reaction conditions typically employ polar aprotic solvents like NMP or DMSO mixed with water, which helps solubilize the inorganic bases such as potassium carbonate or cesium carbonate required to neutralize the acid byproduct. This careful balancing of steric protection and catalytic activation ensures that the reaction proceeds with high conversion and minimal side-product formation.
Impurity control is further reinforced during the workup and purification stages. Unlike the prior art where isomers co-elute or require complex separation, the intermediates in this new route (Compound IIIa and IIIb) possess distinct physical properties that facilitate purification before the final deprotection. The patent data indicates that the final product contains less than 0.15% of the isomer impurity (Compound IX) and non-detectable levels of the self-coupling byproduct (Compound VIII). This level of purity is achieved without resorting to preparative HPLC or flash chromatography, which are bottlenecks in large-scale synthesis. Instead, the process relies on crystallization from solvents like toluene or isopropyl ether. The ability to purge impurities through crystallization is a hallmark of a robust pharmaceutical process, as it provides a definitive "kill step" for genotoxic or structurally similar impurities. For R&D teams, understanding this mechanism validates the scalability of the route, as crystallization is a well-understood unit operation that can be easily modeled and controlled in pilot and commercial plants, ensuring batch-to-batch consistency.
How to Synthesize 3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)aniline Efficiently
The execution of this synthesis requires precise control over reaction parameters to maximize the benefits of the protective group strategy. The process begins with the N-benzylation of 3-bromo-5-trifluoromethylaniline, which can be achieved using benzyl bromide in the presence of a strong base like sodium tert-butoxide, or alternatively via reductive amination with benzaldehyde using sodium borohydride. Following isolation of the protected intermediate, the critical coupling step is performed using copper(I) iodide as the catalyst and a bidentate ligand in a solvent system such as NMP and water at elevated temperatures (120-145°C). The final debenzylation can be tailored to the available infrastructure; catalytic hydrogenation using Pd/C is highly effective and atom-economical, while acidic hydrolysis offers a metal-free alternative. Each step is designed to be telescoped or isolated with minimal loss, ensuring overall process efficiency. For detailed operational parameters, stoichiometry, and specific workup procedures, please refer to the standardized synthesis guide below.
- Perform N-benzylation on 3-bromo-5-trifluoromethylaniline using a benzyl halide or aldehyde to protect the amino group, forming Compound II.
- Execute a copper-catalyzed coupling reaction between the protected intermediate (Compound II) and 4-methyl-1H-imidazole in the presence of a base and ligand to form Compound III.
- Conduct a debenzylation step via hydrogenation or acidic hydrolysis to remove the protecting group, yielding the final high-purity Nilotinib intermediate (Compound IV).
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented synthesis route translates into tangible strategic advantages beyond mere chemical elegance. The primary value driver is the substantial reduction in manufacturing costs derived from the elimination of expensive reagents and complex purification steps. By avoiding the use of diphenyl phosphoryl azide and removing the necessity for column chromatography, the process significantly lowers the Cost of Goods Sold (COGS). Chromatography is a major cost center in fine chemical manufacturing due to high solvent consumption, silica gel costs, and low throughput; replacing it with crystallization drastically improves the process mass intensity (PMI) and throughput capacity. Furthermore, the high selectivity of the reaction means that raw material utilization is optimized, reducing the waste disposal burden associated with off-spec batches. This efficiency allows for more competitive pricing models for the final API, providing a buffer against market volatility in raw material costs.
- Cost Reduction in Manufacturing: The economic impact of this route is profound because it replaces hazardous and costly reagents with commodity chemicals while simplifying the purification train. The avoidance of column chromatography alone represents a massive saving in operational expenditure, as it reduces solvent usage by orders of magnitude and eliminates the need for specialized chromatography columns and silica media. Additionally, the high yield and purity mean that fewer batches are rejected, maximizing the return on investment for every kilogram of starting material purchased. This lean manufacturing approach aligns perfectly with initiatives to reduce waste and energy consumption in pharmaceutical production.
- Enhanced Supply Chain Reliability: From a supply continuity perspective, this method utilizes widely available starting materials and catalysts, reducing the risk of supply disruptions associated with specialty reagents. The robustness of the chemistry ensures consistent batch quality, which minimizes the risk of production delays caused by out-of-specification results. For supply chain planners, this predictability is invaluable, as it allows for tighter inventory management and more accurate forecasting. The ability to produce high-purity intermediates reliably means that downstream API synthesis can proceed without interruption, securing the supply of life-saving medications to patients.
- Scalability and Environmental Compliance: The process is explicitly designed for industrial mass production, featuring conditions that are readily transferable from the laboratory to the pilot plant and finally to commercial scale. The use of standard reactors and the absence of exotic high-pressure or cryogenic conditions simplify equipment requirements. Moreover, the reduction in solvent waste and hazardous reagents supports environmental compliance goals, making it easier to obtain necessary regulatory approvals for manufacturing sites. This sustainability aspect is increasingly important for multinational corporations aiming to meet their carbon footprint and green chemistry targets.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. They are derived from the specific advantages and data points presented in the patent documentation, providing clarity on how this method compares to existing industry standards. Understanding these details is crucial for technical teams evaluating the feasibility of technology transfer and for commercial teams assessing the value proposition of suppliers utilizing this route.
Q: How does this new synthesis method improve impurity profiles compared to direct coupling?
A: By introducing an N-benzyl protecting group prior to the coupling reaction, the method effectively suppresses the formation of self-coupling byproducts (Compound VIII) and significantly reduces the generation of the 5-position isomer impurity (Compound IX) to less than 0.15%, ensuring superior chemical purity over 99.5%.
Q: What are the key advantages for industrial scale-up regarding reagent costs?
A: Unlike previous methods such as WO2004/005281 which rely on expensive and hazardous reagents like diphenyl phosphoryl azide (DPPA) for Curtius rearrangement, this novel route utilizes cost-effective copper catalysis and standard benzylation reagents, eliminating the need for complex purification steps like column chromatography.
Q: Is this process suitable for large-scale commercial production of tyrosine kinase inhibitors?
A: Yes, the patent explicitly states the method is suitable for industrial mass production due to its simple operation, high yield, and robust control over critical quality attributes, making it ideal for the commercial scale-up of complex oncology drug intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Nilotinib Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the synthesis of complex oncology intermediates requires not just chemical expertise but a deep commitment to quality and scalability. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your supply needs are met with precision and reliability. Our facilities are equipped with state-of-the-art rigorous QC labs capable of verifying stringent purity specifications, including the detection of trace isomers and genotoxic impurities at ppm levels. We understand that the integrity of your final API depends on the quality of the building blocks we provide, which is why we adhere to the highest standards of GMP and process safety.
We invite you to collaborate with us to leverage this advanced synthesis technology for your Nilotinib projects. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating how this route can optimize your overall budget. Please contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us help you secure a stable, high-quality supply chain for your critical pharmaceutical intermediates.