Scalable Synthesis of Fluorinated Quinazolinone-Triazole Hybrids for Advanced Pharmaceutical Applications
Scalable Synthesis of Fluorinated Quinazolinone-Triazole Hybrids for Advanced Pharmaceutical Applications
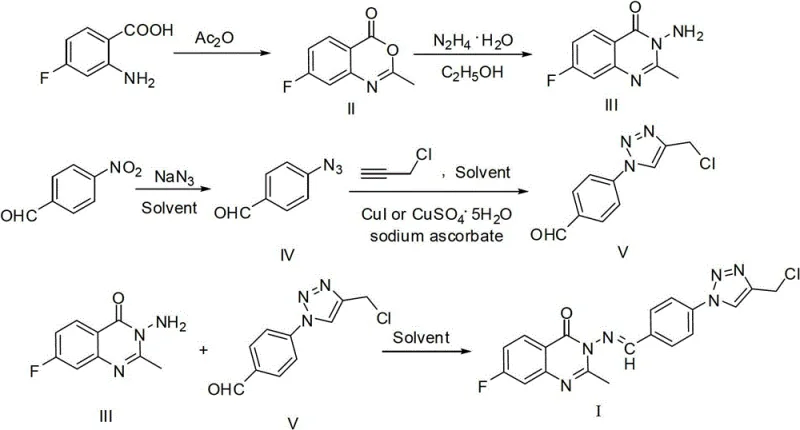
The pharmaceutical landscape is constantly evolving towards more complex heterocyclic scaffolds that offer enhanced biological activity and metabolic stability. Patent CN103059001A introduces a sophisticated yet practical methodology for synthesizing a novel quinazolinone Schiff base containing a 1,2,3-triazole moiety, specifically 3-(4-((4-chloromethyl)-1H-1,2,3-triazole)benzylideneamino-2-methyl-7-fluoro-4(3H)-quinazolinone. This compound represents a strategic convergence of three potent pharmacophores: the quinazolinone core, known for its kinase inhibitory properties; the triazole ring, valued for its bioisosteric capabilities; and the Schiff base linkage, which often contributes to antimicrobial and anticancer activities. For R&D directors and procurement specialists seeking reliable pharmaceutical intermediates supplier partnerships, understanding the nuances of this synthesis is critical. The patent outlines a five-step convergent route that maximizes atom economy while utilizing commodity chemicals, positioning this molecule as a viable candidate for further drug development programs targeting oncology and infectious diseases.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditionally, the synthesis of complex heterocyclic hybrids involving quinazolinones and triazoles has been plagued by inefficiencies that hinder commercial viability. Conventional routes often rely on multi-step linear syntheses where the overall yield drops precipitously with each additional transformation, leading to excessive waste generation and inflated production costs. Many historical methods require harsh reaction conditions, such as strong acids or elevated temperatures exceeding 200°C, which can degrade sensitive functional groups like the chloromethyl moiety essential for downstream derivatization. Furthermore, older protocols frequently utilize stoichiometric amounts of expensive coupling reagents or toxic heavy metal catalysts that are difficult to remove to ppm levels, creating significant bottlenecks in purification and posing environmental compliance risks. These factors collectively result in extended lead times and unreliable supply chains for high-value pharmaceutical intermediates, making it challenging for manufacturers to secure consistent quality at a competitive price point.
The Novel Approach
In stark contrast, the methodology described in patent CN103059001A offers a streamlined, convergent strategy that addresses these historical pain points with elegance and efficiency. By splitting the synthesis into two parallel branches—one constructing the fluorinated quinazolinone core and the other building the triazole-functionalized aldehyde—the process minimizes the longest linear sequence, thereby preserving overall yield. The use of acetic anhydride as both a reagent and solvent in the initial cyclization step eliminates the need for exotic solvents, while the subsequent hydrazinolysis proceeds smoothly in ethanol, a green solvent preferred by modern regulatory standards. The integration of Copper-catalyzed Azide-Alkyne Cycloaddition (CuAAC), commonly known as "Click Chemistry," ensures high regioselectivity and rapid reaction kinetics under mild conditions. This novel approach not only simplifies the operational workflow but also significantly enhances the purity profile of the final product, making it an ideal candidate for cost reduction in pharmaceutical intermediates manufacturing without compromising on structural integrity or biological potential.
Mechanistic Insights into Convergent Heterocyclic Assembly
The chemical elegance of this synthesis lies in its precise control over reaction mechanisms to ensure high fidelity and minimal impurity formation. The first branch begins with the cyclization of 2-amino-4-fluorobenzoic acid with acetic anhydride at 100-140°C. Mechanistically, the amino group attacks the carbonyl of the anhydride, followed by intramolecular nucleophilic attack by the carboxylic acid oxygen onto the activated amide carbonyl, expelling acetate to form the stable 7-fluoro-2-methyl-4(3H)-3,1-benzoxazinone ring. This intermediate is then subjected to hydrazinolysis, where hydrazine hydrate acts as a bidentate nucleophile. It attacks the lactone carbonyl, opening the ring and subsequently cyclizing to form the quinazolinone core with the release of water. This step is crucial as it installs the exocyclic amine necessary for the final Schiff base condensation, and the use of ethanol as a solvent facilitates the precipitation of the product, driving the equilibrium forward.
Simultaneously, the second branch constructs the triazole fragment via a classic nucleophilic aromatic substitution followed by a click reaction. 4-Nitrobenzaldehyde undergoes substitution with sodium azide in polar aprotic solvents like HMPA or DMF at 25-80°C, replacing the nitro group with an azide functionality. This azide then participates in a Cu(I)-catalyzed cycloaddition with 3-chloroallyne. The copper catalyst, generated in situ from CuI or CuSO4 with sodium ascorbate, coordinates with the alkyne to form a copper-acetylide species, which then reacts with the azide to form the 1,4-disubstituted 1,2,3-triazole ring with perfect regiocontrol. Finally, the two branches converge through a condensation reaction between the quinazolinone amine and the triazole aldehyde. This dehydration reaction, typically catalyzed by heat in toluene or ethanol, forms the imine (Schiff base) linkage. The entire pathway is designed to avoid side reactions, ensuring that the chloromethyl group remains intact for potential further functionalization.
How to Synthesize 3-(4-((4-Chloromethyl)-1H-1,2,3-triazole)benzylideneamino-2-methyl-7-fluoro-4(3H)-quinazolinone Efficiently
Executing this synthesis requires careful attention to stoichiometry and temperature control to maximize the reported yields, which range from 89% to 96% for individual steps. The process begins with the preparation of the benzoxazinone intermediate, where maintaining the temperature between 100°C and 140°C is critical to ensure complete cyclization without decomposition. Following isolation, the hydrazinolysis step should be monitored closely by TLC to prevent over-reaction, although the protocol is robust enough to tolerate slight variations. For the triazole formation, the exclusion of oxygen during the click chemistry step is paramount to prevent oxidation of the copper catalyst, which would stall the reaction. The final condensation step benefits from the removal of water, often achieved by using a Dean-Stark trap if toluene is employed, or simply by the crystallization of the product upon cooling in ethanol. Detailed standardized operating procedures for each unit operation are essential for technology transfer.
- Cyclize 2-amino-4-fluorobenzoic acid with acetic anhydride at 100-140°C to form 7-fluoro-2-methyl-4(3H)-3,1-benzoxazinone.
- React the benzoxazinone intermediate with hydrazine hydrate in ethanol at 60-80°C to yield 2-methyl-3-amino-7-fluoro-4(3H)-quinazolinone.
- Convert 4-nitrobenzaldehyde to 4-azidobenzaldehyde using sodium azide in polar aprotic solvents like HMPA or DMF.
- Perform Copper-catalyzed Azide-Alkyne Cycloaddition (CuAAC) between the azide and 3-chloroallyne to form the triazole aldehyde.
- Condense the triazole aldehyde with the quinazolinone amine in toluene or ethanol at reflux to generate the final Schiff base product.
Commercial Advantages for Procurement and Supply Chain Teams
From a commercial perspective, this synthetic route offers compelling advantages that directly address the concerns of procurement managers and supply chain heads regarding cost, reliability, and scalability. The primary driver for cost efficiency is the selection of starting materials; 2-amino-4-fluorobenzoic acid and 4-nitrobenzaldehyde are commodity chemicals available in bulk quantities from multiple global suppliers, mitigating the risk of single-source dependency. The elimination of precious metal catalysts like palladium or platinum in favor of inexpensive copper salts drastically reduces the raw material cost per kilogram. Furthermore, the high yields observed in each step minimize the amount of starting material required to produce a fixed quantity of the final API intermediate, effectively lowering the cost of goods sold (COGS). The use of common solvents such as ethanol, toluene, and ethyl acetate simplifies solvent recovery and recycling processes, contributing to further operational savings and environmental compliance.
- Cost Reduction in Manufacturing: The process achieves significant cost optimization by utilizing a convergent synthesis strategy that reduces the total number of unit operations compared to linear alternatives. By avoiding the use of expensive protecting groups and specialized reagents, the manufacturing overhead is substantially lowered. The high atom economy of the click chemistry step ensures that the majority of the reactant mass is incorporated into the final product, minimizing waste disposal costs. Additionally, the simplicity of the workup procedures, which rely on filtration and crystallization rather than complex chromatographic separations for bulk purification, reduces labor and equipment time, leading to a more economical production process overall.
- Enhanced Supply Chain Reliability: The reliance on widely available, non-proprietary raw materials ensures a stable and resilient supply chain capable of withstanding market fluctuations. Since the synthesis does not depend on exotic reagents with long lead times, production schedules can be maintained with greater predictability. The robustness of the reaction conditions, which tolerate a range of temperatures and solvent grades, allows for flexibility in sourcing and manufacturing locations. This flexibility is crucial for maintaining continuous supply to downstream pharmaceutical customers, reducing the risk of production stoppages due to material shortages or logistical delays.
- Scalability and Environmental Compliance: The synthetic route is inherently scalable, having been demonstrated effectively from gram to multi-kilogram scales in the patent examples. The reactions proceed under moderate pressures and temperatures, reducing the engineering requirements for specialized high-pressure reactors. From an environmental standpoint, the process generates less hazardous waste compared to traditional methods, as it avoids the use of heavy metals and toxic solvents where possible. The ability to recycle solvents like ethanol and toluene further aligns the process with green chemistry principles, facilitating easier regulatory approval and reducing the environmental footprint of large-scale manufacturing operations.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the production and application of this quinazolinone-triazole hybrid. These insights are derived directly from the experimental data and theoretical framework provided in the patent documentation, offering clarity on process feasibility and product quality. Understanding these details is essential for stakeholders evaluating the potential of this compound for inclusion in their drug discovery pipelines or manufacturing portfolios.
Q: What are the key advantages of this synthetic route for quinazolinone derivatives?
A: The patented route utilizes readily available starting materials like 2-amino-4-fluorobenzoic acid and employs mild reaction conditions. It avoids complex purification steps by leveraging high-yield crystallization and standard chromatography, ensuring a robust process suitable for industrial scale-up.
Q: How does the incorporation of the triazole moiety affect the biological potential?
A: The 1,2,3-triazole ring serves as a bioisostere that enhances metabolic stability and binding affinity through hydrogen bonding and dipole interactions. Combining this with the quinazolinone scaffold creates a multi-target pharmacophore with demonstrated antitumor activity against cell lines such as A-549 lung carcinoma.
Q: Is the copper catalyst used in the click chemistry step difficult to remove?
A: The process utilizes standard copper catalysts like CuI or CuSO4·5H2O with sodium ascorbate. The workup procedure involves simple filtration to remove insolubles followed by aqueous washing and organic extraction, effectively reducing residual metal content to meet stringent pharmaceutical specifications without requiring specialized scavengers.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Quinazolinone Schiff Base Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of translating innovative patent chemistry into commercial reality. Our team of expert chemists possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to pilot plant and finally to full-scale manufacturing is seamless and efficient. We are committed to delivering high-purity pharmaceutical intermediates that meet stringent purity specifications, utilizing our rigorous QC labs to verify every batch against the highest industry standards. Our facility is equipped to handle the specific requirements of heterocyclic synthesis, including the safe handling of azides and the precise control of exothermic reactions, guaranteeing both safety and quality.
We invite you to collaborate with us to leverage this advanced synthetic technology for your next-generation therapeutic programs. By partnering with NINGBO INNO PHARMCHEM, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality needs. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us help you accelerate your development timeline and reduce your manufacturing costs with our proven expertise in complex organic synthesis and supply chain management.