Scalable One-Pot Decarboxylation Strategy for High-Purity Aryl Amidine Intermediates
Introduction to Advanced Aryl Amidine Synthesis Technology
The landscape of fine chemical manufacturing is constantly evolving, driven by the need for more efficient, sustainable, and cost-effective synthetic routes for critical pharmaceutical intermediates. A significant breakthrough in this domain is documented in Chinese Patent CN110963943B, which discloses a novel method for synthesizing aryl amidine compounds through a one-pot decarboxylation reaction. This technology represents a paradigm shift from traditional multi-step protocols, offering a streamlined pathway that utilizes readily available starting materials such as nitrones and aryl isocyanates. For R&D directors and procurement strategists, this patent offers a compelling value proposition: the ability to produce high-purity aryl amidines under mild, controllable conditions with exceptional reaction yields. By leveraging this intellectual property, manufacturers can significantly reduce process complexity while maintaining stringent quality standards required for downstream drug synthesis.
The core innovation lies in the direct transformation of nitrone and isocyanate precursors into the desired amidine scaffold without the need for isolating unstable intermediates. This approach not only minimizes waste generation but also drastically shortens the production cycle time. As the global demand for heterocyclic compounds in medicinal chemistry continues to surge, having access to a robust, scalable synthesis method for aryl amidines becomes a critical competitive advantage. This report analyzes the technical merits of this patented process, evaluating its potential to redefine cost structures and supply chain reliability for reliable aryl amidine supplier networks globally.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of amidines has been fraught with challenges that hinder large-scale commercial adoption. Traditional methodologies, such as those reported by Schaefer in 1961 or Weintraub in 2000, often rely on the nucleophilic addition of substituted nitriles or the O-alkylation of secondary amides. These classical routes frequently necessitate harsh reaction conditions, including the use of strong acids like hydrogen chloride or specialized reagents such as boron tetrafluoride triethoxy salts. Furthermore, many existing protocols involve multi-step sequences where intermediates like ethylimidate salts must be isolated and purified before proceeding to the final aminolysis step. This fragmentation of the synthetic route inherently increases material loss, extends lead times, and escalates the overall cost of goods sold (COGS).
Additionally, conventional methods often suffer from limited substrate scope and sensitivity to functional groups, restricting their utility in the synthesis of complex pharmaceutical intermediates. The requirement for specialized catalysts, such as N-acetylcysteine in methanol at elevated temperatures, further complicates the process economics due to catalyst recovery and disposal costs. From a supply chain perspective, reliance on these inefficient pathways introduces volatility, as any disruption in the supply of niche reagents can halt production entirely. Consequently, the industry has long sought a direct, efficient synthesis method that bypasses these bottlenecks while delivering high purity and yield.
The Novel Approach
The methodology outlined in Patent CN110963943B addresses these historical pain points through an elegant one-pot decarboxylation strategy. By reacting a nitrone compound (Compound 1) with an aryl isocyanate (Compound 2) in the presence of a specific catalytic system, the process directly affords the target aryl amidine in a single operational step. This eliminates the need for isolating the oxadiazolidinone intermediate, which is typically formed via a [1,3]-dipolar cycloaddition, and instead promotes its immediate conversion to the amidine through base-catalyzed rearrangement and carbon dioxide extrusion. The simplicity of this approach is underscored by the use of common organic solvents and inexpensive inorganic bases.
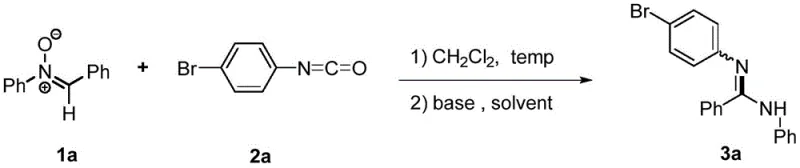
As illustrated in the reaction scheme above, the transformation proceeds smoothly under mild conditions, typically at room temperature (25°C) to moderate heating (up to 80°C), with reaction times around 12 hours. Experimental data from the patent demonstrates that switching the solvent to ethanol (EtOH) and utilizing cesium carbonate (Cs2CO3) as the base catalyst can drive yields as high as 93%, a substantial improvement over the 0-47% yields observed with other solvent-base combinations like toluene or DBU. This robustness makes the process highly attractive for cost reduction in pharmaceutical intermediates manufacturing, as it maximizes atom economy and minimizes downstream purification burdens.
Mechanistic Insights into Nitrone-Isocyanate Cycloaddition and Decarboxylation
To fully appreciate the technical sophistication of this method, one must understand the underlying mechanistic pathway. The reaction initiates with a [1,3]-dipolar cycloaddition between the nitrone dipole and the isocyanate dipolarophile. The nitrone, characterized by its N-O double bond and electron-rich oxygen, attacks the electrophilic carbon of the isocyanate group (N=C=O). This step forms a five-membered oxadiazolidinone ring intermediate. While previous literature, such as the work by Anderson in 2014, described the isolation of similar intermediates followed by styryl transfer, the present invention optimizes the conditions to facilitate the immediate collapse of this ring.
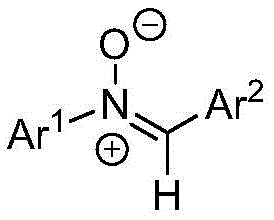
The presence of the base catalyst, specifically cesium carbonate, plays a pivotal role in the subsequent decarboxylation step. The base likely deprotonates a transient species or stabilizes the transition state required for the ring-opening of the oxadiazolidinone. This leads to the extrusion of carbon dioxide (CO2) and the formation of the stable C=N double bond characteristic of the amidine functionality. The use of dichloromethane (CH2Cl2) as a co-catalyst or additive, as specified in the patent claims, further modulates the reaction environment, potentially enhancing the solubility of intermediates or stabilizing charged species during the rearrangement. This mechanistic clarity allows for precise control over impurity profiles, ensuring that side reactions such as polymerization or hydrolysis are minimized.
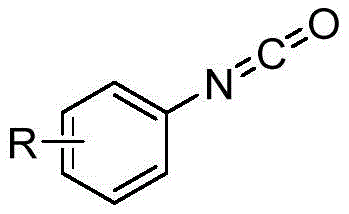
Furthermore, the tolerance of various substituents on both the nitrone (Ar1, Ar2) and the isocyanate (R) rings demonstrates the versatility of this mechanism. Electron-donating or electron-withdrawing groups, including halogens (Cl, Br), alkyls, and trifluoromethyl groups, are well-tolerated without significant erosion of yield. For instance, the synthesis of 4-chlorophenyl and 4-trifluoromethylphenyl derivatives proceeded with yields exceeding 89%, indicating that the electronic nature of the aromatic rings does not inhibit the cycloaddition-decarboxylation cascade. This broad substrate scope is crucial for R&D teams aiming to generate diverse libraries of high-purity aryl amidines for structure-activity relationship (SAR) studies.
How to Synthesize Aryl Amidines Efficiently
Implementing this synthesis route in a laboratory or pilot plant setting requires adherence to specific operational parameters to ensure reproducibility and safety. The process begins with the dissolution of the nitrone and aryl isocyanate substrates in a polar protic solvent like ethanol, which has been identified as the optimal medium for maximizing conversion. The addition of the catalytic system must be performed under an inert atmosphere, typically nitrogen, to prevent moisture ingress which could hydrolyze the sensitive isocyanate starting material. Following the reaction period, a standard aqueous workup involving extraction with dichloromethane and drying over anhydrous magnesium sulfate effectively isolates the crude product.
- Dissolve N-alpha-diphenylnitrone and the selected aryl isocyanate in a suitable organic solvent such as ethanol or toluene at room temperature.
- Add dichloromethane (CH2Cl2) and a basic catalyst like cesium carbonate (Cs2CO3) to the reaction mixture under nitrogen protection.
- Stir the mixture for approximately 12 hours, monitor by TLC, then extract with dichloromethane and purify via silica gel column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this patented technology translates into tangible strategic benefits beyond mere chemical efficiency. The primary advantage lies in the drastic simplification of the supply chain for raw materials. Unlike legacy methods that require specialized, hard-to-source reagents, this process relies on commodity chemicals like aryl isocyanates and nitrones, which are widely available from multiple global vendors. This diversification of the supplier base mitigates the risk of single-source dependency and ensures business continuity even during market fluctuations. Moreover, the elimination of transition metal catalysts removes the need for expensive and time-consuming heavy metal scavenging steps, which are often a regulatory bottleneck in API manufacturing.
- Cost Reduction in Manufacturing: The economic impact of this one-pot method is profound. By consolidating multiple synthetic steps into a single operation, manufacturers can significantly reduce labor costs, energy consumption, and solvent usage. The ability to run the reaction at room temperature (25°C) rather than requiring cryogenic conditions or high-pressure reactors further lowers utility expenses. Additionally, the high yields (up to 93%) mean less raw material is wasted, directly improving the gross margin of the final product. The avoidance of complex purification techniques like preparative HPLC, favoring standard silica gel chromatography instead, ensures that the cost of goods remains competitive for large-scale production.
- Enhanced Supply Chain Reliability: The robustness of the reaction conditions contributes to a more predictable manufacturing schedule. Since the process is tolerant to a wide range of functional groups and operates under mild conditions, the risk of batch failure due to thermal runaway or sensitivity issues is minimized. This reliability allows for tighter inventory management and shorter lead times for customers. The use of ethanol as a preferred solvent also aligns with green chemistry principles, reducing the environmental footprint and simplifying waste disposal compliance, which is increasingly critical for maintaining operating licenses in regulated jurisdictions.
- Scalability and Environmental Compliance: Scaling this process from gram to kilogram or tonne levels is straightforward due to the absence of hazardous reagents and extreme conditions. The generation of carbon dioxide as the only stoichiometric byproduct simplifies waste stream management compared to processes generating heavy metal sludge or toxic organic waste. This environmental compatibility facilitates faster regulatory approval for new drug applications (NDAs) where the synthesis route of the intermediate is scrutinized. Consequently, partners adopting this technology can position themselves as sustainable leaders in the commercial scale-up of complex pharmaceutical intermediates.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this decarboxylation technology. These insights are derived directly from the experimental data and claims presented in the patent documentation, providing a factual basis for decision-making. Understanding these nuances helps stakeholders evaluate the feasibility of integrating this method into their existing production portfolios.
Q: What are the key advantages of this one-pot decarboxylation method over traditional amidine synthesis?
A: Unlike traditional multi-step methods requiring harsh conditions or special catalysts, this patent describes a mild, one-pot process at room temperature using commercially available nitrones and isocyanates, achieving yields up to 93% with simplified purification.
Q: Which solvents and catalysts provide the optimal yield for aryl amidine production?
A: Experimental data indicates that ethanol (EtOH) serves as the superior solvent compared to toluene or acetonitrile, while cesium carbonate (Cs2CO3) acts as the most effective base catalyst, significantly outperforming potassium carbonate or organic bases like DBU.
Q: Is this synthesis method scalable for industrial manufacturing of pharmaceutical intermediates?
A: Yes, the process utilizes simple raw materials, operates at ambient temperatures reducing energy costs, and avoids complex transition metal catalysts, making it highly suitable for commercial scale-up and consistent supply chain reliability.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Aryl Amidines Supplier
The technological advancements detailed in Patent CN110963943B underscore the immense potential of modern organic synthesis to drive efficiency in the fine chemical sector. At NINGBO INNO PHARMCHEM, we recognize the value of such innovations and have integrated similar cutting-edge methodologies into our CDMO capabilities. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from lab-scale discovery to industrial manufacturing is seamless. We maintain stringent purity specifications and operate rigorous QC labs to guarantee that every batch of aryl amidines meets the exacting standards required by the global pharmaceutical industry.
We invite potential partners to leverage our expertise to optimize their supply chains and reduce time-to-market for critical drug candidates. By collaborating with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific project needs. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments. Let us demonstrate how our commitment to innovation and quality can support your long-term strategic goals in the competitive landscape of pharmaceutical intermediates.