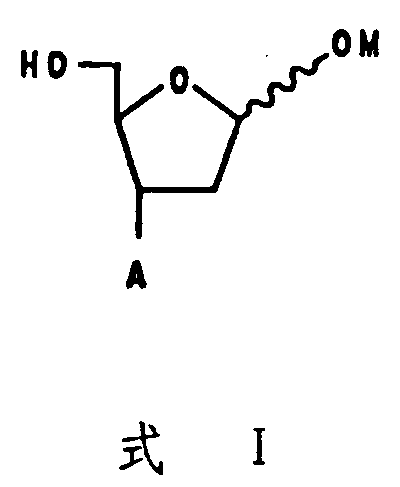
Scalable Asymmetric Synthesis of 3-Substituted Furanosides for Antiviral Drug Manufacturing
The pharmaceutical industry's relentless pursuit of effective antiretroviral therapies has placed a premium on the efficient synthesis of nucleoside analogs, particularly those exhibiting potent reverse transcriptase inhibitory activity. Patent CN1066658A introduces a groundbreaking methodology for the asymmetric synthesis of 3-substituted furanose and furanoside compounds, which serve as critical intermediates in the manufacture of life-saving medications such as AZT and FLT for HIV treatment. This technology represents a significant paradigm shift from traditional carbohydrate chemistry, moving away from the laborious modification of natural sugars towards a more convergent and stereocontrolled assembly of the furanose core. By leveraging advanced organometallic catalysis and regioselective transformations, this patent outlines a pathway that not only simplifies the molecular construction but also enhances the overall purity profile required for stringent pharmaceutical applications. The ability to introduce diverse functional groups at the 3-prime position with high fidelity opens new avenues for developing next-generation antiviral agents with improved pharmacokinetic properties.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the preparation of 3-prime substituted nucleosides has relied heavily on the direct modification of existing 2-prime deoxynucleosides or the complex manipulation of naturally occurring pentoses like D-xylose. These conventional pathways are often plagued by inherent inefficiencies, including the necessity for extensive protecting group strategies that add multiple synthetic steps and reduce overall atom economy. The steric hindrance present in rigid sugar rings frequently leads to poor regioselectivity during substitution reactions, resulting in difficult-to-separate isomeric mixtures that compromise the final API purity. Furthermore, the reliance on expensive chiral pool starting materials limits the scalability and cost-effectiveness of these processes, creating bottlenecks in the supply chain for high-volume antiviral production. The multi-step nature of these traditional routes also increases the exposure of sensitive intermediates to harsh conditions, potentially leading to degradation and lower yields that are unacceptable for commercial manufacturing standards.
The Novel Approach
In stark contrast, the methodology disclosed in CN1066658A utilizes a linear, acyclic strategy that builds the furanose ring from simple, commercially available precursors such as propargyl alcohol and allyl halides. This approach bypasses the structural constraints of natural sugars, allowing chemists to install the critical 3-prime substituent with precise stereochemical control before the ring closure occurs. The use of asymmetric epoxidation followed by titanium-mediated nucleophilic ring opening provides a robust mechanism for establishing the desired chirality without the need for resolution steps. This streamlined process significantly reduces the number of unit operations required, thereby minimizing solvent usage and waste generation while improving the overall throughput. By decoupling the stereochemical setup from the ring formation, this novel route offers unparalleled flexibility in generating a wide array of 3-substituted derivatives, making it an ideal platform for both process development and large-scale commercial synthesis of complex nucleoside analogs.
Mechanistic Insights into Titanium-Mediated Asymmetric Epoxidation and Ring Opening
The core innovation of this synthesis lies in the sophisticated application of Sharpless asymmetric epoxidation to generate chiral epoxy alcohols with exceptional enantiomeric excess. In this critical step, a catalytic system composed of titanium tetraisopropoxide and a chiral tartrate ester, such as diisopropyl tartrate, directs the oxidation of the allylic alcohol substrate using tert-butyl hydroperoxide. The coordination geometry of the titanium center ensures that oxygen delivery occurs from a specific face of the olefin, establishing the absolute configuration of the resulting epoxide with high fidelity. This stereochemical information is then faithfully transmitted through subsequent transformations, ensuring that the final furanoside product maintains the required optical purity for biological activity. The rigorous control of reaction parameters, including temperature maintenance at minus 20 degrees Celsius and the use of activated molecular sieves to exclude moisture, is paramount to achieving the reported levels of selectivity and yield in this catalytic cycle.
Following the establishment of chirality, the process employs a unique titanium-based nucleophilic reagent system to effect regioselective ring opening of the epoxide intermediate. Reagents of the general formula Ti(OR prime)nA4-minus n act as mild yet effective Lewis acids that activate the epoxide towards nucleophilic attack by species such as fluoride, azide, or thiobenzoate. This activation lowers the energy barrier for ring opening and directs the nucleophile to the desired carbon center, avoiding the formation of unwanted regioisomers that typically plague acid-catalyzed openings. The versatility of this titanium reagent system allows for the introduction of various functional groups at the 3-position simply by altering the ligand A, providing a modular approach to library synthesis. This mechanistic elegance ensures that the synthetic route remains robust across different substrate variations, maintaining high conversion rates and simplifying downstream purification processes.
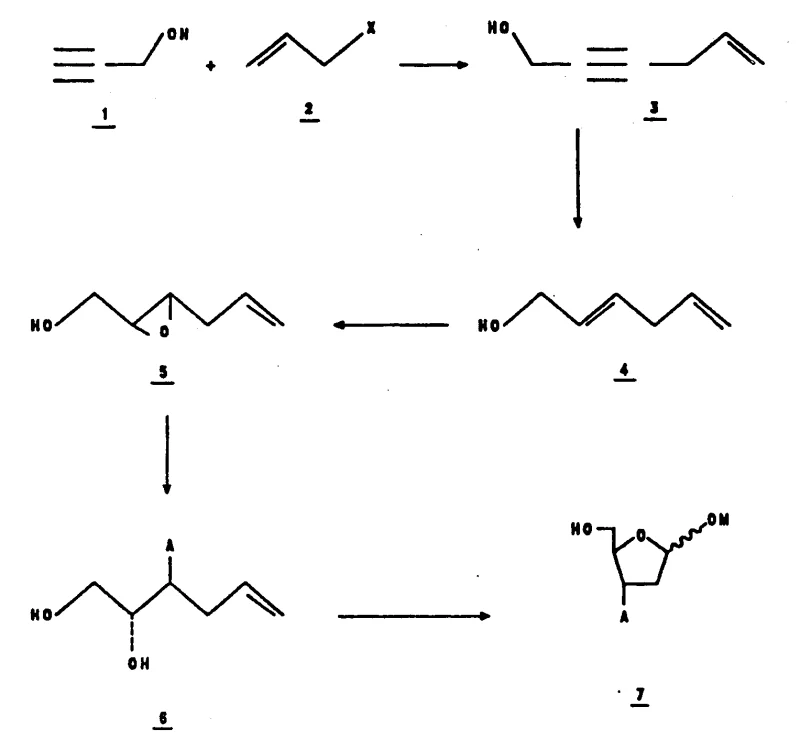
How to Synthesize 3-Substituted Furanoside Efficiently
The synthesis of these high-value intermediates begins with the condensation of propargyl alcohol and allyl halides under basic aqueous conditions to form an enyne intermediate, which is subsequently reduced to a dienol. This dienol serves as the substrate for the asymmetric epoxidation step, where careful control of stoichiometry and aging time of the catalyst mixture is essential for optimal performance. Following epoxidation, the crude epoxy alcohol is treated with the specific titanium nucleophile to install the 3-prime substituent, followed by ozonolysis to cleave the terminal olefin and reveal the aldehyde functionality required for ring closure. The final cyclization and glycosylation are achieved through acid-catalyzed conditions in methanol, yielding the target furanoside as a mixture of anomers that can be separated or utilized directly depending on the downstream coupling requirements. Detailed standardized synthesis steps are provided below for technical reference.
- Condensation of propargyl alcohol with allyl halides to form enyne intermediates.
- Asymmetric epoxidation using titanium tartrate catalysts to establish chirality.
- Regioselective nucleophilic ring opening followed by ozonolysis and glycosylation.
Commercial Advantages for Procurement and Supply Chain Teams
From a strategic procurement perspective, the adoption of this synthesis route offers substantial advantages in terms of raw material security and cost structure optimization. The reliance on bulk commodity chemicals like propargyl alcohol and allyl chloride eliminates the dependency on volatile agricultural sugar markets, ensuring a more stable and predictable supply chain for key starting materials. The reduction in synthetic step count directly correlates to lower operational expenditures, as fewer reactors, less solvent, and reduced labor hours are required to produce the same quantity of finished intermediate. Additionally, the high regioselectivity of the titanium-mediated steps minimizes the formation of difficult-to-remove impurities, which reduces the burden on quality control laboratories and decreases the risk of batch rejection due to specification failures. These factors combine to create a manufacturing process that is not only economically superior but also more resilient to market fluctuations and supply disruptions.
- Cost Reduction in Manufacturing: The elimination of expensive chiral pool starting materials and the reduction of protection-deprotection sequences lead to significant cost savings in raw material procurement and waste disposal. By streamlining the synthesis to fewer high-yielding steps, manufacturers can achieve a lower cost of goods sold without compromising on the quality or purity of the final antiviral intermediate. The use of catalytic amounts of chiral inducers rather than stoichiometric chiral auxiliaries further enhances the economic viability of the process, making it competitive for generic drug production where margin pressure is intense.
- Enhanced Supply Chain Reliability: Utilizing widely available petrochemical-derived building materials ensures that production schedules are not held hostage by seasonal variations or crop failures associated with natural sugar sources. The robustness of the reaction conditions allows for flexible manufacturing campaigns, enabling suppliers to respond rapidly to surges in demand for HIV therapeutics. Furthermore, the simplified purification protocols reduce the lead time required for batch release, facilitating faster inventory turnover and improving cash flow dynamics for pharmaceutical partners relying on just-in-time delivery models.
- Scalability and Environmental Compliance: The process is designed with scale-up in mind, utilizing reaction conditions that are easily managed in large-scale stainless steel reactors without requiring exotic high-pressure or cryogenic equipment beyond standard chilling capabilities. The improved atom economy and reduced solvent intensity align with modern green chemistry principles, helping manufacturers meet increasingly stringent environmental regulations and sustainability goals. This eco-friendly profile enhances the corporate social responsibility standing of the supply chain, making it an attractive option for multinational corporations committed to reducing their carbon footprint.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this patented synthesis technology. These answers are derived directly from the experimental data and claims within the patent documentation to ensure accuracy and reliability for decision-makers. Understanding these nuances is critical for evaluating the feasibility of integrating this route into existing manufacturing portfolios.
Q: What is the primary advantage of this synthesis route over conventional sugar modification?
A: This route avoids complex protection-deprotection sequences on sugar rings, utilizing simpler acyclic precursors that allow for better stereocontrol and fewer processing steps.
Q: How is high enantiomeric excess achieved in this process?
A: High enantiomeric excess is secured through the Sharpless asymmetric epoxidation step, utilizing chiral tartrate esters and titanium catalysts to dictate the stereochemistry early in the synthesis.
Q: Is this method suitable for large-scale production of HIV drug intermediates?
A: Yes, the use of readily available starting materials like propargyl alcohol and robust reaction conditions makes this methodology highly adaptable for commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 3-Substituted Furanoside Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of securing a reliable supply of high-quality antiviral intermediates to support the global fight against infectious diseases. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the sophisticated chemistry described in CN1066658A can be translated into robust industrial processes. We maintain stringent purity specifications and operate rigorous QC labs equipped with state-of-the-art analytical instrumentation to guarantee that every batch of 3-substituted furanoside meets the exacting standards required for pharmaceutical grade applications. Our commitment to technical excellence ensures that potential scale-up risks are identified and mitigated early in the development phase.
We invite procurement leaders and R&D directors to collaborate with us on optimizing their supply chains for nucleoside analogs. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into how implementing this specific synthesis route can impact your overall budget and timeline. We encourage you to contact our technical procurement team to索取 specific COA data and route feasibility assessments tailored to your project needs. Let us partner with you to accelerate the delivery of life-saving medications to patients worldwide through superior chemical manufacturing.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →