Advanced Synthesis of Pyrrolopyrimidine Intermediates for Scalable JAK Inhibitor Manufacturing
Advanced Synthesis of Pyrrolopyrimidine Intermediates for Scalable JAK Inhibitor Manufacturing
The pharmaceutical landscape for Janus Kinase (JAK) inhibitors continues to evolve rapidly, driven by the critical need for effective treatments in myelofibrosis and rheumatoid arthritis. Central to the production of next-generation therapeutics like Rucotinib and Baricitinib is the availability of high-quality heterocyclic building blocks. A recent technological breakthrough, documented in patent CN109651424B, introduces a robust and economically viable synthesis method for 7-protecting group-4-(1-hydrogen-pyrazol-4-yl)pyrrolo[2,3-d]pyrimidine. This compound serves as a pivotal intermediate in the value chain of these life-saving medications. By shifting away from traditional, resource-intensive pathways, this innovation addresses long-standing bottlenecks in purity and cost, positioning itself as a cornerstone for reliable pharmaceutical intermediate supplier networks globally.
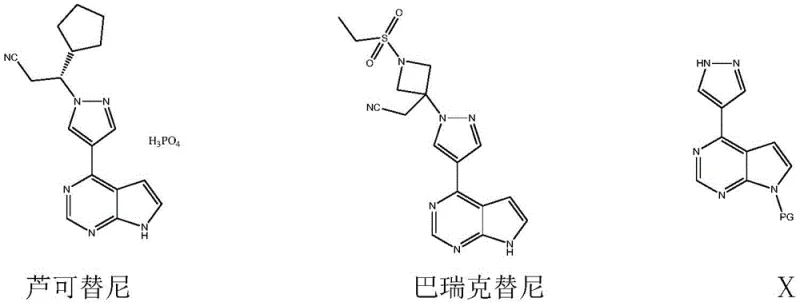
The structural complexity of JAK inhibitors demands precise synthetic strategies to ensure regulatory compliance and therapeutic efficacy. As illustrated in the molecular diagrams, the core pyrrolopyrimidine scaffold linked to a pyrazole ring is a non-negotiable structural motif for biological activity. The ability to construct this scaffold efficiently determines the commercial viability of the final drug substance. Consequently, mastering the synthesis of this specific intermediate is not merely a chemical challenge but a strategic imperative for supply chain resilience in the oncology and immunology sectors.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the preparation of this key intermediate has relied on convoluted multi-step sequences that impose significant burdens on manufacturing efficiency and cost structures. Conventional routes, such as those disclosed in earlier patents like US20100190981, typically initiate with the formylation and chlorination of dihydroxypyrimidine, followed by amination and Wittig olefination. These steps require harsh reagents and generate substantial chemical waste. Furthermore, the subsequent cyclization and protection steps often necessitate the use of expensive transition metal catalysts, specifically tetrakis(triphenylphosphine)palladium, for Suzuki coupling reactions to install the pyrazole moiety.
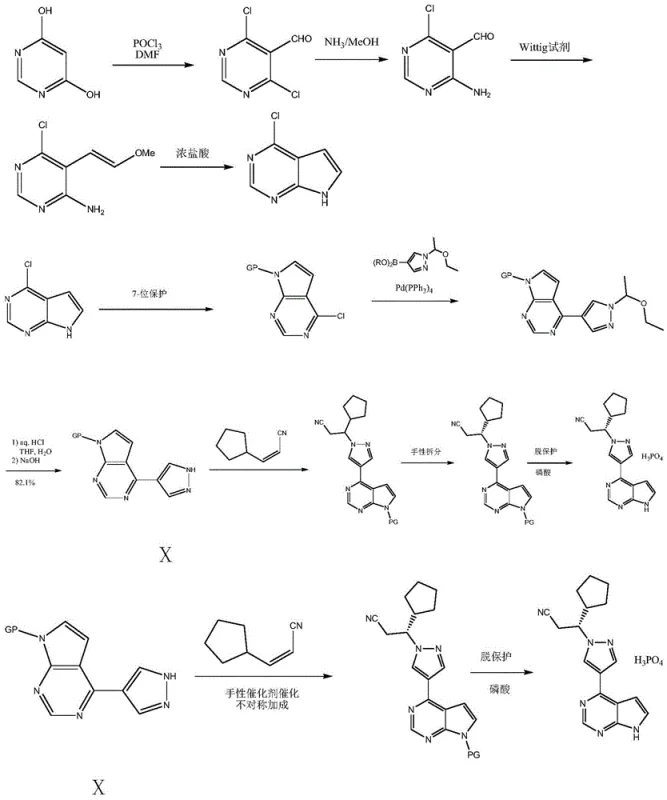
The reliance on palladium-catalyzed cross-coupling introduces severe complications regarding heavy metal residue control, necessitating additional purification stages that erode overall yield. Moreover, the requirement for specialized boronic acid esters, which themselves require multi-step synthesis involving Grignard reagents or metal lithiation, exacerbates the cost profile. From a procurement perspective, these dependencies create vulnerability to raw material price volatility and supply disruptions. The cumulative effect is a process with low atom economy, high environmental impact, and operational complexity that hinders seamless commercial scale-up of complex pharmaceutical intermediates.
The Novel Approach
In stark contrast, the methodology outlined in CN109651424B represents a paradigm shift towards simplicity and sustainability. This novel approach bypasses the need for precious metal catalysis and exotic starting materials entirely. Instead, it leverages readily available commodity chemicals such as cyanoacetoacetate and haloacetaldehyde acetals to construct the carbon skeleton. The strategy employs a clever sequence of dehydrohalogenation and condensation reactions to build the pyrrolopyrimidine core directly. This fundamental redesign of the synthetic tree eliminates the most costly and hazardous steps of the prior art, offering a direct pathway to cost reduction in API manufacturing.
By utilizing a protecting group strategy that is compatible with mild reaction conditions, the new process ensures high selectivity and minimizes the formation of difficult-to-remove impurities. The elimination of column chromatography in favor of crystallization-based purification further enhances its suitability for industrial implementation. This streamlined workflow not only reduces the physical footprint required for production but also significantly shortens the cycle time from raw material intake to finished intermediate. For supply chain heads, this translates to enhanced reliability and the ability to respond more agilely to market demand fluctuations without compromising on quality standards.
Mechanistic Insights into the Condensation and Cyclization Strategy
The heart of this innovative synthesis lies in the efficient construction of the heterocyclic system through controlled condensation mechanisms. The process initiates with the reaction between cyanoacetoacetate and a haloacetaldehyde acetal diol in the presence of a base. This dehydrohalogenation step generates a reactive intermediate that undergoes intramolecular cyclization upon treatment with formamidine hydrochloride. The precise control of temperature, typically maintained between 20°C and 45°C during these stages, is critical to preventing side reactions and ensuring the formation of the desired 2-(7-hydropyrrolo[2,3-d]pyrimidin-4-yl)acetate structure with high fidelity.
Following the core formation, the synthesis proceeds through a sophisticated functionalization sequence involving protection, methylation, and ring closure. The amino group at the 7-position is protected using reagents like 2-(chloromethoxy)ethyl trimethylsilane, which provides stability during subsequent transformations. The introduction of the pyrazole ring is achieved via methylation with DMFDMA followed by condensation with hydrazine hydrate. This specific sequence allows for the regioselective formation of the pyrazole moiety attached to the pyrrolopyrimidine core. The final steps involve chlorination of the hydroxyl group followed by catalytic hydrogenation dechlorination, a clean reduction method that removes the chlorine atom without affecting other sensitive functional groups, yielding the target intermediate with exceptional purity.
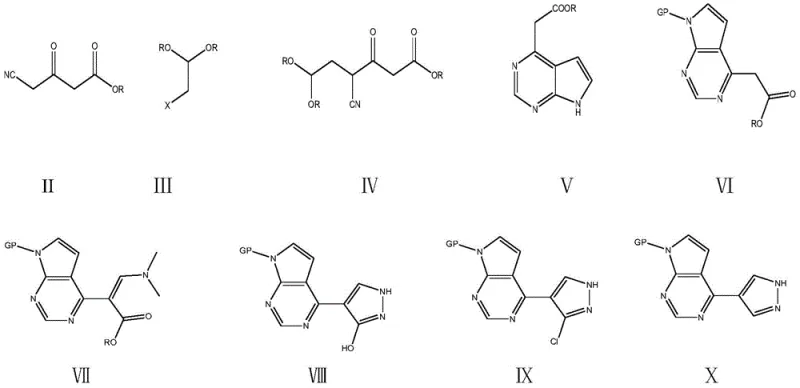
Impurity control is inherently built into this mechanistic design. The use of crystallization as the primary purification method at multiple stages, rather than chromatographic separation, indicates that the reaction profiles are clean and the byproducts are structurally distinct enough to be removed physically. The patent data reports liquid phase purities consistently exceeding 99% across various examples, demonstrating the robustness of the chemical pathway. For R&D directors, this level of purity is paramount as it simplifies the downstream processing of the final API, reducing the risk of genotoxic impurities or heavy metal contamination that often plague processes involving transition metal catalysts.
How to Synthesize 7-Protecting Group-4-(1H-pyrazol-4-yl)pyrrolo[2,3-d]pyrimidine Efficiently
Implementing this synthesis requires careful attention to reaction parameters such as solvent selection, base strength, and temperature gradients to maximize yield and safety. The process is designed to be operationally simple, utilizing common solvents like DMF and THF which are easily sourced and recycled in an industrial setting. The following guide outlines the standardized operational framework derived from the patent examples, providing a clear roadmap for technical teams looking to adopt this superior manufacturing protocol. Detailed standard operating procedures for each unit operation are essential to replicate the high success rates reported in the intellectual property documentation.
- Perform dehydrohalogenation of cyanoacetoacetate and haloacetaldehyde acetal, followed by condensation with formamidine hydrochloride to form the pyrrolopyrimidine core.
- Protect the amino group, methylate using DMFDMA, and condense with hydrazine hydrate to construct the pyrazole ring.
- Chlorinate the hydroxypyrazole intermediate and perform catalytic hydrogenation dechlorination to yield the final protected intermediate.
Commercial Advantages for Procurement and Supply Chain Teams
The transition to this novel synthesis route offers profound economic and logistical benefits that extend far beyond the laboratory bench. For procurement managers, the most immediate impact is the drastic simplification of the raw material portfolio. By replacing expensive, custom-synthesized reagents like Wittig salts and palladium catalysts with bulk commodities like cyanoacetoacetate and hydrazine, the direct material costs are significantly reduced. This shift mitigates the risk associated with sourcing specialized chemicals from limited suppliers, thereby enhancing the overall security of the supply chain against geopolitical or market-driven disruptions.
- Cost Reduction in Manufacturing: The elimination of precious metal catalysts removes the need for expensive scavenging resins and rigorous heavy metal testing, which are costly line items in API production budgets. Furthermore, the high atom economy of the condensation reactions means less raw material is wasted as byproduct, directly improving the cost-per-kilogram metric. The ability to perform one-pot operations for multiple steps reduces solvent consumption and energy usage associated with isolation and drying between stages, leading to substantial operational expenditure savings without compromising product quality.
- Enhanced Supply Chain Reliability: Dependence on complex, multi-step precursor synthesis is a known vulnerability in pharmaceutical supply chains. This new method utilizes starting materials that are widely available from multiple global vendors, ensuring consistent availability and competitive pricing. The robustness of the reaction conditions, which tolerate a range of temperatures and do not require stringent anhydrous environments for every step, reduces the likelihood of batch failures due to minor process deviations. This reliability is crucial for maintaining continuous production schedules and meeting strict delivery commitments to downstream API manufacturers.
- Scalability and Environmental Compliance: As regulatory pressure mounts regarding pharmaceutical waste and environmental impact, this green chemistry approach offers a distinct advantage. The reduction in hazardous waste generation and the avoidance of toxic heavy metals simplify wastewater treatment protocols and lower disposal costs. The process is inherently scalable, moving smoothly from kilogram to ton-scale production without the engineering challenges associated with exothermic metal-catalyzed couplings. This scalability ensures that the supply can grow in tandem with the clinical and commercial success of the final drug products, supporting long-term business growth.
Frequently Asked Questions (FAQ)
Understanding the technical nuances of this synthesis is vital for stakeholders evaluating its adoption for commercial production. The following questions address common inquiries regarding the practical implementation, quality attributes, and strategic benefits of this patented technology. These insights are derived directly from the experimental data and claims within the patent specification, providing a factual basis for decision-making processes regarding technology transfer and vendor qualification.
Q: What are the key advantages of the new synthesis route over conventional methods?
A: The new route eliminates expensive Wittig reagents and palladium catalysts, utilizing cheap raw materials like cyanoacetoacetate and achieving higher atom economy with simplified one-pot operations.
Q: What purity levels can be achieved with this manufacturing process?
A: The patented method consistently delivers liquid phase purity exceeding 99%, with total yields reaching over 72.2%, ensuring high-quality standards for downstream API synthesis.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process features stable reaction conditions, low waste generation, and avoids complex chromatography, making it highly favorable for green industrial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 7-Protecting Group-4-(1H-pyrazol-4-yl)pyrrolo[2,3-d]pyrimidine Supplier
The successful translation of this patented synthesis from laboratory concept to commercial reality requires a partner with deep technical expertise and proven manufacturing capabilities. NINGBO INNO PHARMCHEM stands at the forefront of this domain, possessing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our facilities are equipped with state-of-the-art reactors and rigorous QC labs capable of meeting the stringent purity specifications demanded by top-tier pharmaceutical companies. We understand that consistency is key, and our quality management systems are designed to ensure every batch meets the highest international standards for safety and efficacy.
We invite global partners to collaborate with us to leverage this advanced synthesis technology for their JAK inhibitor programs. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage potential clients to reach out for specific COA data and route feasibility assessments to verify how this innovative process can optimize your supply chain and reduce overall manufacturing costs. Let us be your trusted ally in navigating the complexities of modern pharmaceutical intermediate production.