Optimizing PARP Inhibitor Production: A Novel Seven-Step Synthetic Route for Industrial Scale-Up
Introduction to Patent CN114437085A
The pharmaceutical landscape for oncology treatments continues to evolve, driven by the demand for efficient synthesis of complex small molecules like Rucaparib, a potent PARP inhibitor used in treating advanced ovarian cancer. A critical bottleneck in the supply chain has always been the production of its key precursor, 8-fluoro-1,3,4,5-tetrahydro-6H-azepino[5,4,3-cd]indol-6-one (Formula II). Patent CN114437085A, published in May 2022, introduces a transformative preparation method that reconstructs the synthetic logic by prioritizing the formation of the benzolactam ring before the indole ring. This strategic reversal not only simplifies the reaction sequence but also utilizes inexpensive, readily available raw materials such as p-fluorobenzaldehyde. For R&D directors and procurement specialists, this patent represents a significant leap forward in process chemistry, offering a pathway that mitigates safety hazards associated with explosive reagents while ensuring high yields and exceptional purity suitable for GMP manufacturing environments.
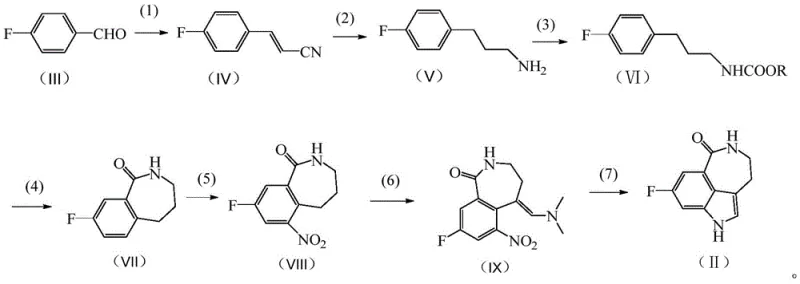
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this innovation, the industry relied on three primary synthetic routes, each plagued by distinct economic and safety deficiencies that hindered scalable production. As illustrated in the comparative analysis of existing technologies, Synthesis Scheme 1 depended heavily on 1-dimethylamino-2-nitroethylene, a reagent that is not only prohibitively expensive but also results in disappointingly low yields during the critical reduction steps from compound 7 to compound 8. Similarly, Synthesis Scheme 2 suffered from poor efficiency in the transformation of compound 6 to compound 11, necessitating the use of costly trifluoroacetic acid (TFA) and triethylsilane (TES), which drastically inflated the cost of goods sold. Perhaps most critically, Synthesis Scheme 3 utilized nitromethane, a substance with known explosive properties that poses severe safety risks in large-scale industrial reactors, alongside a lengthy reaction sequence that cumulatively resulted in low overall throughput. These legacy methods created substantial barriers for reliable pharmaceutical intermediate suppliers aiming to secure consistent inventory levels.
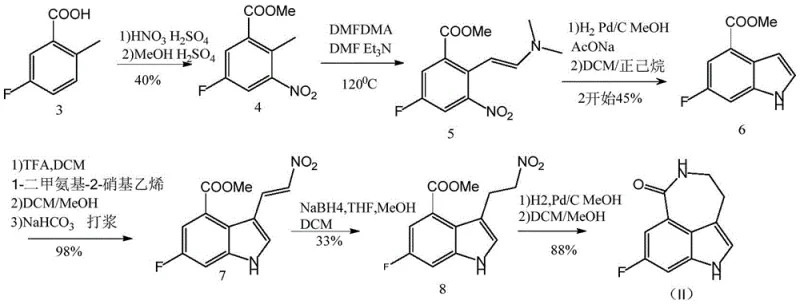
The Novel Approach
The methodology disclosed in CN114437085A fundamentally re-engineers the synthesis by adopting a linear, seven-step progression that begins with the condensation of p-fluorobenzaldehyde and acetonitrile. This approach constructs the seven-membered benzolactam ring early in the sequence via a robust dehydration cyclization, followed by the formation of the indole moiety in the final stages. By avoiding the use of explosive nitromethane and expensive specialized reagents, the new route achieves a total yield of approximately 39.4% in optimized examples, with product purity consistently reaching 99.4% after simple pulping purification. This streamlined architecture allows for milder reaction conditions, typically operating between 0°C and 140°C, which reduces energy consumption and equipment stress. For procurement managers, this translates to a more resilient supply chain capable of cost reduction in pharmaceutical intermediates manufacturing without compromising on the rigorous quality standards required for oncology drugs.
Mechanistic Insights into the Seven-Step Cascade Synthesis
The core of this technological breakthrough lies in the precise control of reaction kinetics and chemoselectivity across the seven distinct transformations. The process initiates with an alkaline condensation where p-fluorobenzaldehyde reacts with acetonitrile in the presence of a phase transfer catalyst, such as dodecyl trimethyl ammonium chloride, to generate p-fluorocinnamonitrile (Formula IV) with high efficiency. Subsequent catalytic hydrogenation using skeletal nickel or palladium-carbon under moderate pressure (2-3 MPa) selectively reduces the nitrile group to a primary amine (Formula V) without affecting the aromatic fluorine substituent. The protection of this amine via reaction with chloroformates forms a carbamate intermediate (Formula VI), which serves as the crucial precursor for the ring-closing step. The cyclization is mediated by strong dehydrating agents like polyphosphoric acid (PPA) or phosphorus oxychloride, effectively closing the seven-membered lactam ring to yield Formula VII. This sequence demonstrates superior atom economy compared to prior art, minimizing waste generation and simplifying downstream processing.
Following the construction of the lactam core, the synthesis proceeds through a controlled nitration using mixed acid (nitric and sulfuric acid) at low temperatures (0-10°C) to install the nitro group at the ortho position, yielding Formula VIII. The introduction of the dimethylaminomethylene group via reaction with N,N-dimethylformamide dimethyl acetal (DMFDMA) creates an enamine intermediate (Formula IX) that is primed for the final cyclization. The concluding step involves a reductive cyclization, achievable either through catalytic hydrogenation or chemical reduction with iron powder, which simultaneously reduces the nitro group and closes the indole ring to form the target molecule (Formula II). This mechanistic pathway ensures that impurities are minimized at each stage, particularly through the use of specific solvent systems like dichloromethane and methanol for purification, resulting in a high-purity API intermediate that requires minimal further refinement.
How to Synthesize 8-fluoro-1,3,4,5-tetrahydro-6H-azepino[5,4,3-cd]indol-6-one Efficiently
Implementing this synthesis requires strict adherence to the temperature profiles and stoichiometric ratios defined in the patent to maximize yield and safety. The process is designed to be scalable, moving seamlessly from laboratory glassware to industrial reactors by managing exothermic events during the nitration and cyclization phases. Operators must ensure precise control of hydrogen pressure during the reduction steps and maintain anhydrous conditions where necessary to prevent hydrolysis of sensitive intermediates. The detailed standardized synthesis steps, including specific work-up procedures and purification protocols, are outlined below to guide technical teams in replicating this high-efficiency route.
- Condense p-fluorobenzaldehyde with acetonitrile in alkaline solution to form p-fluorocinnamonitrile.
- Perform catalytic hydrogenation on the nitrile to generate 3-(4-fluorophenyl)propylamine.
- React the amine with chloroformate to protect the nitrogen, followed by dehydration cyclization to construct the benzolactam ring.
- Execute nitration, enamine formation with DMFDMA, and final reductive cyclization to yield the target indole-fused lactam.
Commercial Advantages for Procurement and Supply Chain Teams
For supply chain leaders, the adoption of this patented process offers immediate strategic benefits by addressing the volatility often associated with complex heterocyclic synthesis. The shift away from hazardous and expensive reagents directly impacts the bottom line by stabilizing raw material costs and reducing the need for specialized safety infrastructure. Furthermore, the robustness of the reaction conditions means that production schedules are less likely to be disrupted by batch failures or safety incidents, ensuring a steady flow of materials for downstream API production. This reliability is paramount for maintaining continuity in the global supply of oncology medications.
- Cost Reduction in Manufacturing: The elimination of high-cost reagents such as 1-dimethylamino-2-nitroethylene and the avoidance of low-yield transformation steps significantly lowers the variable cost per kilogram. By utilizing commodity chemicals like p-fluorobenzaldehyde and acetonitrile, manufacturers can decouple their production costs from the fluctuating prices of specialty fine chemicals. Additionally, the simplified purification process, which relies on standard solvent pulping rather than complex chromatography, reduces solvent consumption and waste disposal expenses, leading to substantial overall cost savings.
- Enhanced Supply Chain Reliability: The reliance on widely available starting materials mitigates the risk of supply shortages that often plague niche chemical markets. Since the route avoids explosive substances like nitromethane, it faces fewer regulatory hurdles regarding storage and transport, allowing for faster logistics and reduced lead times. This accessibility ensures that manufacturers can scale production rapidly to meet market demand without being constrained by the availability of exotic precursors.
- Scalability and Environmental Compliance: The mild reaction conditions and the absence of highly toxic or explosive intermediates make this process inherently safer and easier to scale from pilot plant to commercial tonnage. The reduced generation of hazardous waste aligns with increasingly stringent environmental regulations, lowering the compliance burden on manufacturing facilities. This green chemistry approach not only protects the environment but also enhances the corporate sustainability profile of the supply chain partners involved.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel synthetic route. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on how this method outperforms traditional approaches in terms of safety, yield, and operational feasibility.
Q: How does this new process improve safety compared to prior art?
A: Unlike previous methods that utilize explosive nitromethane or expensive unstable reagents, this patented route employs stable, commercially available starting materials like p-fluorobenzaldehyde and avoids hazardous high-energy intermediates, significantly reducing industrial safety risks.
Q: What is the expected purity profile of the intermediate produced via this method?
A: The process includes specific purification steps, such as stirring and pulping in solvents like dichloromethane and methanol, which consistently deliver product purity exceeding 99%, meeting stringent requirements for downstream API synthesis.
Q: Why is this route considered more cost-effective for large-scale manufacturing?
A: The route eliminates the need for costly reagents like 1-dimethylamino-2-nitroethylene and avoids low-yield steps found in conventional schemes. The use of common catalysts like Raney Nickel or Pd/C and mild reaction conditions further drives down operational expenditures.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Rucaparib Intermediate Supplier
As the global demand for PARP inhibitors continues to rise, securing a stable source of high-quality intermediates is critical for pharmaceutical developers. NINGBO INNO PHARMCHEM leverages extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production to bring this advanced synthesis to life. Our state-of-the-art facilities are equipped to handle the specific catalytic hydrogenation and cyclization steps required by this patent, ensuring stringent purity specifications are met through our rigorous QC labs. We understand that consistency is key in oncology drug development, and our commitment to process optimization guarantees a supply of 8-fluoro-1,3,4,5-tetrahydro-6H-azepino[5,4,3-cd]indol-6-one that meets the highest international standards.
We invite procurement directors and R&D teams to collaborate with us to evaluate the feasibility of integrating this cost-effective route into your supply chain. By contacting our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your volume requirements. We are prepared to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can support your long-term production goals while minimizing risk and maximizing efficiency.