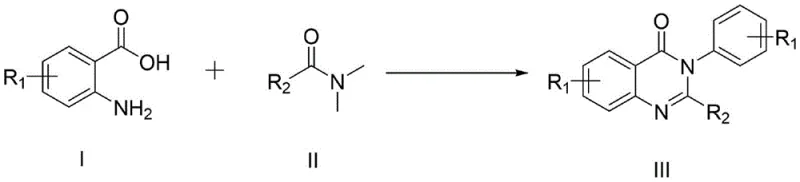
Scalable Synthesis of 4-(3H)-Quinazolinone Derivatives for Commercial API Production
The pharmaceutical industry continuously seeks robust synthetic pathways for nitrogen-containing heterocycles, particularly the 4-(3H)-quinazolinone scaffold which serves as a critical core structure for numerous bioactive agents including kinase inhibitors and antifungal drugs. Patent CN114230526B introduces a transformative one-pot methodology that utilizes imidazole hydrochloride as a novel organocatalyst to facilitate the condensation of anthranilic acid derivatives with DMF derivatives. This approach addresses long-standing challenges in the field by avoiding the use of hazardous isocyanates and corrosive mineral acids that have traditionally plagued the synthesis of these valuable pharmaceutical intermediates. By leveraging a metal-free catalytic system, this technology offers a cleaner reaction profile that aligns perfectly with modern green chemistry principles and stringent regulatory requirements for residual impurities in active pharmaceutical ingredients. The strategic implementation of this synthesis route provides a compelling opportunity for manufacturers to enhance their portfolio of high-purity pharmaceutical intermediates while mitigating environmental liabilities associated with heavy metal waste streams.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the quinazolinone core has relied heavily on the Aza-Wittig reaction involving phosphinimines and isocyanates, a pathway that introduces significant toxicity concerns due to the highly reactive and hazardous nature of isocyanate reagents. Alternative strategies employing 4H-3,1-benzoxazin-4-one intermediates often suffer from multi-step sequences that drastically reduce overall throughput and increase production costs through excessive solvent consumption and purification burdens. Furthermore, direct cyclization using formamide typically requires excessively high temperatures that lead to reactant carbonization and the formation of difficult-to-remove tarry byproducts, severely compromising the isolation yield and purity of the final API intermediate. The reliance on organic metal catalysts in some literature precedents further complicates the manufacturing process by necessitating expensive scavenging steps to meet strict ppm limits for heavy metals in drug substances. These cumulative inefficiencies create substantial bottlenecks in the supply chain, driving up the cost of goods sold and extending lead times for critical oncology and cardiovascular medication precursors.
The Novel Approach
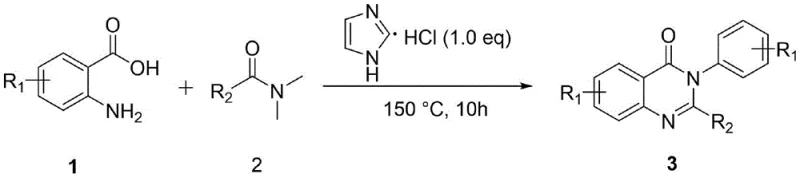
In stark contrast, the innovative method disclosed in the patent data utilizes a simple yet highly effective imidazole hydrochloride catalytic system that operates under remarkably mild conditions relative to the thermal stability of the products. This one-pot synthesis eliminates the need for dichloromethane and concentrated hydrochloric acid, thereby removing two of the most problematic solvents and reagents from the manufacturing workflow in terms of occupational health and environmental compliance. The process does not require specialized high-pressure autoclaves or inert gas protection, allowing for execution in standard glass-lined or stainless steel reactors that are readily available in most fine chemical production facilities. By streamlining the reaction into a single thermal step at 150°C, the methodology significantly reduces energy consumption and operator handling time compared to multi-stage traditional protocols. This operational simplicity translates directly into enhanced process reliability and scalability, making it an ideal candidate for the commercial scale-up of complex pharmaceutical intermediates required for global drug supply chains.
Mechanistic Insights into Imidazole Hydrochloride-Catalyzed Cyclization
The catalytic efficacy of imidazole hydrochloride in this transformation stems from its unique ability to activate the carbonyl functionality of the DMF derivative while simultaneously stabilizing the transition state during the cyclization event. Mechanistic studies suggest that the acidic proton of the imidazolium salt facilitates the nucleophilic attack of the anthranilic amine group, promoting the formation of the key amide intermediate without inducing excessive hydrolysis of the solvent. Crucially, the presence of this specific organocatalyst suppresses the competing transamidation side reactions that typically degrade yield in non-catalyzed thermal condensations of similar substrates. Optimization experiments revealed that maintaining a precise catalyst loading of 0.5 equivalents is essential to balance reaction kinetics with product stability, as deviations can lead to incomplete conversion or increased impurity profiles. This fine-tuned mechanistic pathway ensures that the electronic properties of the substituents on the aromatic rings are accommodated effectively, allowing for the successful synthesis of diverse 2,3-disubstituted quinazolinone derivatives with consistent quality attributes.
Impurity control is a paramount concern for R&D directors overseeing the development of oncology intermediates like those used for Idelalisib, and this catalytic system offers distinct advantages in managing the杂质 spectrum. The absence of transition metals eliminates the risk of metal-complexed impurities that are notoriously difficult to purge during crystallization, thereby simplifying the analytical validation process for regulatory filings. Furthermore, the reaction demonstrates good functional group tolerance for electron-donating substituents such as methyl and methoxy groups, which remain intact throughout the thermal cycle without undergoing unwanted oxidation or rearrangement. Although electron-withdrawing groups like nitro or fluoro substituents present greater challenges, the robustness of the imidazole hydrochloride system still allows for the isolation of target compounds where other methods might fail completely. The resulting crude product profiles are generally cleaner, requiring less aggressive chromatographic purification and enabling more efficient recrystallization protocols to achieve the stringent purity specifications demanded by international pharmacopoeias.
How to Synthesize 4-(3H)-Quinazolinone Efficiently
To implement this synthesis effectively in a laboratory or pilot plant setting, operators must adhere to specific thermal and stoichiometric parameters defined in the patent examples to ensure reproducible outcomes. The general procedure involves charging a reaction vessel with the anthranilic acid derivative, the DMF derivative, and the imidazole hydrochloride catalyst, followed by heating to 150°C for a duration of approximately 10 hours. Detailed standardized synthesis steps see the guide below.
- Combine anthranilic acid derivative, DMF derivative, and 0.5 equivalents of imidazole hydrochloride catalyst in a reaction vessel.
- Heat the mixture to 150°C in an oil bath and stir for 10 hours without requiring inert gas protection.
- Quench with water, extract with ethyl acetate, wash, dry, and purify via silica gel chromatography to isolate the target quinazolinone.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, the adoption of this metal-free synthetic route offers substantial cost savings by eliminating the need for expensive palladium or copper catalysts and the associated ligand systems that drive up raw material expenses. The reliance on commercially available bulk chemicals such as DMF and imidazole hydrochloride ensures a stable supply chain with minimal risk of vendor lock-in or geopolitical sourcing disruptions that often affect specialized reagents. Additionally, the removal of dichloromethane from the process reduces the regulatory burden and disposal costs associated with halogenated solvent waste, contributing to a lower total cost of ownership for the manufacturing site. These factors combine to create a more resilient supply chain capable of sustaining long-term production campaigns for high-volume API intermediates without frequent process interruptions or raw material shortages.
- Cost Reduction in Manufacturing: The elimination of transition metal catalysts removes the necessity for costly metal scavenging resins and extensive washing procedures, directly lowering the variable cost per kilogram of the produced intermediate. By operating at atmospheric pressure without the need for high-pressure autoclaves, the process reduces capital depreciation costs and maintenance expenses associated with complex reactor systems. The simplified workup procedure involving basic aqueous quenching and extraction minimizes solvent usage and labor hours, further enhancing the overall economic efficiency of the manufacturing campaign. These cumulative efficiencies allow for a more competitive pricing structure when supplying high-purity pharmaceutical intermediates to cost-sensitive generic drug manufacturers.
- Enhanced Supply Chain Reliability: Utilizing commodity chemicals like anthranilic acid derivatives and DMF ensures that raw material availability is not constrained by niche supplier capacity or complex import/export regulations. The robustness of the reaction conditions means that production can be easily transferred between different manufacturing sites without requiring specialized equipment modifications or extensive re-validation efforts. This flexibility significantly reduces lead time for high-purity pharmaceutical intermediates by allowing for parallel production runs across multiple facilities to meet surging market demand. Consequently, supply chain managers can maintain higher safety stock levels with confidence, knowing that the synthesis route is dependable and less prone to failure due to reagent quality variations.
- Scalability and Environmental Compliance: The absence of toxic isocyanates and corrosive acids simplifies the environmental permitting process and reduces the risk of accidental exposure incidents during large-scale operations. Waste streams generated from this process are easier to treat and neutralize compared to those containing heavy metals or halogenated byproducts, aligning with increasingly strict global environmental sustainability mandates. The thermal stability of the reaction mixture allows for safe scale-up from kilogram to tonne quantities without exothermic runaway risks, ensuring consistent batch-to-batch quality. This environmental and operational safety profile makes the technology highly attractive for contract development and manufacturing organizations seeking to expand their green chemistry capabilities.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this quinazolinone synthesis technology in industrial settings. These answers are derived directly from the experimental data and beneficial effects reported in the patent documentation to ensure accuracy and relevance for decision-makers. Understanding these nuances is critical for evaluating the feasibility of integrating this route into existing production pipelines for oncology and cardiovascular drug intermediates.
Q: What are the primary advantages of this quinazolinone synthesis method over traditional routes?
A: This method eliminates the need for toxic isocyanates and corrosive concentrated hydrochloric acid found in conventional Aza-Wittig or formamide pathways. It operates under metal-free conditions using imidazole hydrochloride, significantly reducing heavy metal contamination risks and simplifying downstream purification processes for pharmaceutical applications.
Q: Does this process require high-pressure equipment or inert gas protection?
A: No, the protocol described in patent CN114230526B specifically notes that the reaction proceeds efficiently at atmospheric pressure without the need for autoclaves or nitrogen gas shielding. This operational simplicity lowers capital expenditure requirements for reactor infrastructure and enhances overall process safety in large-scale manufacturing environments.
Q: What is the functional group tolerance for substrates in this catalytic system?
A: The system demonstrates excellent tolerance for electron-donating groups such as methyl and methoxy substituents on the benzene ring, yielding products with moderate to good efficiency. However, strong electron-withdrawing groups like nitro or fluoro substituents may result in lower conversion rates or reaction failure, necessitating careful substrate selection during route scouting.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 4-(3H)-Quinazolinone Supplier
As a leading CDMO partner, NINGBO INNO PHARMCHEM possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from benchtop discovery to full-scale manufacturing. Our facility is equipped with state-of-the-art rigorous QC labs capable of verifying stringent purity specifications for complex heterocyclic intermediates, guaranteeing that every batch meets the exacting standards required for clinical and commercial use. We understand the critical nature of supply continuity for life-saving medications and have established robust contingency plans to mitigate any potential disruptions in the raw material supply chain. Our technical team is ready to collaborate closely with your R&D department to optimize this specific imidazole-catalyzed route for your unique target molecules.
We invite you to contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. By engaging with us early in your development cycle, you can secure specific COA data and route feasibility assessments that will accelerate your regulatory filing timelines. Let us demonstrate how our expertise in catalytic synthesis can become a strategic asset in your global supply chain, delivering both value and reliability for your most critical pharmaceutical projects.