Revolutionizing Afuresertib Production: A Streamlined Chiral Synthesis for Commercial Scale-Up
Revolutionizing Afuresertib Production: A Streamlined Chiral Synthesis for Commercial Scale-Up
The pharmaceutical landscape for oncology treatments is constantly evolving, with the demand for potent AKT kinase inhibitors like Afuresertib driving significant innovation in process chemistry. Patent CN111620860B, published in early 2023, introduces a groundbreaking preparation method that fundamentally restructures the synthetic approach to this critical therapeutic agent. Unlike traditional pathways that rely heavily on cumbersome protection group strategies, this novel methodology leverages a direct chiral induction strategy via hydrazonation and catalytic cyanation. This technical breakthrough not only simplifies the molecular construction but also addresses the persistent challenges of yield loss and operational complexity associated with multi-step protection cycles. For industry stakeholders, this represents a pivotal shift towards more efficient, green chemistry-compliant manufacturing protocols.
The core innovation lies in the strategic assembly of the chiral amine fragment, which is the pharmacophore responsible for the drug's biological activity. By utilizing 3-fluorophenylacetaldehyde as a starting material and reacting it with a specific chiral auxiliary, (2R)-2-methoxymethyl-1-amino-tetrahydropyrrole, the process establishes the critical stereocenter early in the synthesis. This approach bypasses the need for resolving racemic mixtures later in the pipeline, a common bottleneck in API production. The subsequent transformation involves a titanium-mediated cyanation that locks in the stereochemistry with high fidelity, followed by a clean hydrogenation step. This sequence demonstrates a sophisticated understanding of organometallic catalysis applied to complex pharmaceutical intermediate synthesis, offering a robust alternative to legacy methods.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
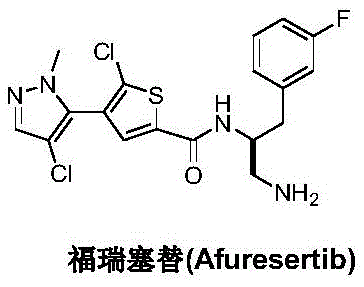
Prior art, specifically referenced in international patent WO2008098104, outlines a synthesis pathway that, while functional, is plagued by inherent inefficiencies suitable only for laboratory-scale discovery rather than commercial manufacturing. The conventional route relies on the synthesis of Intermediate A, which necessitates the use of (2S)-2-amino-3-(3-fluorophenyl)propionic acid as a raw material. To manipulate the functional groups without affecting the sensitive amine, the process requires an initial amino protection step, typically using Boc anhydride. Following this, the carboxyl group must be reduced to an alcohol, and subsequently converted into a phthalimide derivative to reintroduce nitrogen functionality. This sequence involves at least four distinct chemical transformations just to prepare the amine fragment, each carrying a risk of yield erosion and impurity generation.
Furthermore, the reliance on phthalimide chemistry introduces additional purification burdens. The removal of the phthalimide protecting group often requires harsh hydrazine treatment or acidic hydrolysis, which can compromise the integrity of the fluorophenyl moiety or lead to racemization of the chiral center. These competing reactions between adjacent amino groups, as noted in the background analysis of the patent, force chemists to employ repetitive protection and deprotection cycles. From a supply chain perspective, this translates to longer lead times, higher consumption of solvents and reagents, and increased waste generation. The cumulative effect is a process that is economically unviable for large-scale production, failing to meet the rigorous cost and environmental standards required by modern pharmaceutical supply chains.
The Novel Approach
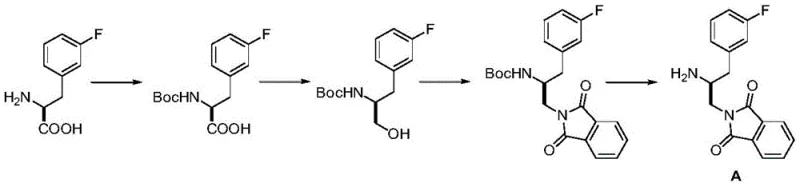
In stark contrast, the methodology disclosed in CN111620860B offers a streamlined, convergent synthesis that drastically reduces the step count and operational complexity. The new route initiates with a hydrazonation reaction between 3-fluorophenylacetaldehyde and the chiral tetrahydropyrrole auxiliary. This condensation is performed under mild thermal conditions (80-85°C) in benzene or toluene, utilizing a water separator to drive the equilibrium forward, ensuring high conversion rates without the need for exotic catalysts. The resulting imine intermediate is then subjected to a highly stereoselective cyanation reaction. By employing titanium tetrachloride as a Lewis acid catalyst at cryogenic temperatures (-78°C), the process achieves precise control over the nucleophilic attack of trimethylsilyl cyanide, establishing the (S)-configuration with exceptional purity.
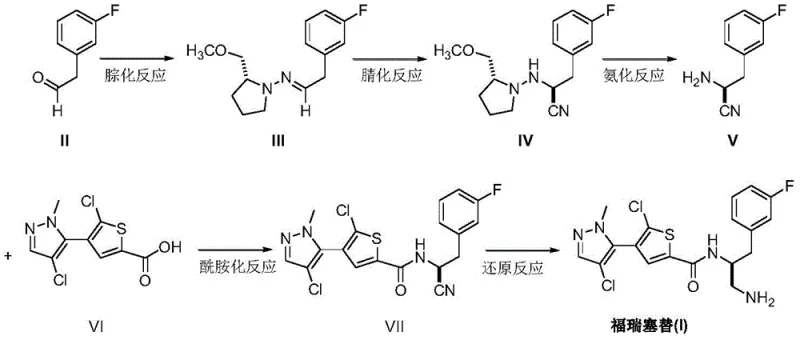
Following the establishment of the chiral nitrile, the auxiliary is removed via a straightforward catalytic hydrogenation using palladium on carbon, yielding the free chiral amine nitrile directly. This fragment is then coupled with the thiophene-carboxylic acid derivative via a standard amidation reaction using thionyl chloride. The final step involves the reduction of the nitrile group to the primary amine using a sodium borohydride and nickel dichloride system. This specific reduction protocol is superior to traditional lithium aluminum hydride reductions as it operates under much milder conditions (25-30°C) and offers better safety profiles for industrial handling. The elimination of the Boc protection/deprotection cycle alone represents a massive leap in process efficiency, directly addressing the yield losses and waste issues inherent in the older technology.
Mechanistic Insights into Titanium-Catalyzed Asymmetric Cyanation
The heart of this synthetic innovation is the titanium-catalyzed asymmetric cyanation step, which serves as the stereo-determining event for the entire molecule. In this mechanism, titanium tetrachloride (TiCl4) acts as a potent Lewis acid, coordinating with the nitrogen atom of the imine intermediate formed in the previous step. This coordination activates the imine carbon towards nucleophilic attack while simultaneously organizing the transition state geometry through the chiral environment provided by the tetrahydropyrrole auxiliary. The bulky methoxymethyl group on the auxiliary creates a steric shield that directs the incoming trimethylsilyl cyanide (TMSCN) to attack exclusively from one face of the planar imine bond. This facial selectivity is crucial for obtaining the desired (S)-enantiomer, which is essential for the biological activity of Afuresertib as an AKT inhibitor.
From an impurity control perspective, this mechanism offers significant advantages over resolution-based approaches. In resolution processes, the maximum theoretical yield is capped at 50% unless dynamic kinetic resolution is employed, and the unwanted enantiomer becomes a difficult-to-remove impurity. Here, the asymmetric induction ensures that the formation of the wrong enantiomer is kinetically suppressed at the source. Furthermore, the use of TiCl4 helps to suppress side reactions such as polymerization of the aldehyde or hydrolysis of the imine, which are common pitfalls in imine chemistry. The subsequent hydrogenation step is equally critical; the use of Pd/C allows for the cleavage of the C-N bond connecting the auxiliary without reducing the nitrile group or the fluorophenyl ring, demonstrating excellent chemoselectivity. This precision minimizes the formation of des-fluoro byproducts or over-reduced amines, ensuring a high-purity profile for the downstream amidation.
How to Synthesize Afuresertib Efficiently
The synthesis of Afuresertib via this patented route requires strict adherence to anhydrous conditions and temperature controls, particularly during the cyanation phase. The process begins with the preparation of the chiral imine, followed by the low-temperature addition of the titanium catalyst and cyanide source. Detailed operational parameters, including specific solvent choices like dichloromethane for the cyanation and ethanol for the final reduction, are critical for reproducibility. The following guide outlines the standardized synthesis steps derived from the patent examples, ensuring optimal yield and purity for pilot and commercial batches.
- Perform hydrazonation of 3-fluorophenylacetaldehyde with a chiral tetrahydropyrrole auxiliary to form the imine intermediate.
- Execute asymmetric cyanation using trimethylsilyl cyanide and titanium tetrachloride at low temperature (-78°C) to establish chirality.
- Conduct catalytic hydrogenation to remove the auxiliary, followed by amidation with the thiophene acid and final nitrile reduction.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this novel synthesis route translates into tangible strategic advantages beyond mere technical elegance. The primary benefit is the drastic simplification of the supply chain for raw materials. By eliminating the need for protected amino acids like Boc-phenylalanine derivatives and avoiding the use of phthalic anhydride for protection, the process relies on more commodity-grade starting materials such as 3-fluorophenylacetaldehyde and simple chiral auxiliaries. This shift reduces dependency on specialized, high-cost building blocks that are often subject to market volatility and long lead times. Consequently, this enhances supply chain resilience, allowing for more flexible sourcing strategies and reducing the risk of production stoppages due to raw material shortages.
- Cost Reduction in Manufacturing: The economic impact of this process is driven by the significant reduction in unit operations. By removing the protection and deprotection steps, the manufacturing timeline is shortened, leading to lower labor costs and reduced utility consumption (heating, cooling, and stirring time). Furthermore, the avoidance of expensive reagents like Boc anhydride and the reduction in solvent usage for intermediate isolations contribute to substantial cost savings. The switch from hazardous reducing agents like LiAlH4 to the safer NaBH4/NiCl2 system also lowers waste disposal costs and insurance premiums associated with handling pyrophoric materials, optimizing the overall cost of goods sold (COGS) for the API.
- Enhanced Supply Chain Reliability: The robustness of the new route ensures consistent quality and delivery performance. The mild reaction conditions, particularly the ambient temperature hydrogenation and reduction steps, reduce the likelihood of thermal runaways or equipment failures that can disrupt production schedules. Additionally, the high stereoselectivity of the cyanation step minimizes the need for costly and time-consuming chiral chromatography or recrystallization purifications to remove enantiomeric impurities. This reliability allows supply chain planners to forecast production outputs with greater accuracy, ensuring a steady flow of high-purity intermediates to downstream formulation teams.
- Scalability and Environmental Compliance: From an environmental, health, and safety (EHS) perspective, this process aligns perfectly with green chemistry principles. The reduction in step count inherently reduces the E-factor (mass of waste per mass of product), lowering the burden on waste treatment facilities. The use of ethanol and methanol as solvents in the final steps replaces more toxic chlorinated solvents often found in older routes, facilitating easier solvent recovery and recycling. This environmental optimization not only ensures compliance with increasingly stringent global regulations but also enhances the corporate sustainability profile, a key metric for modern pharmaceutical partnerships.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on the feasibility and advantages of the new route for potential licensees and manufacturing partners.
Q: How does the new synthesis route improve upon the conventional WO2008098104 method?
A: The new route eliminates the need for multiple amino protection and deprotection steps (such as Boc groups) required in the conventional method, significantly shortening the synthetic sequence and improving overall yield.
Q: What catalysts are used to ensure high enantiomeric purity in this process?
A: The process utilizes a chiral tetrahydropyrrole auxiliary combined with titanium tetrachloride (TiCl4) during the cyanation step to strictly control stereochemistry at the alpha-carbon.
Q: Is this synthesis method suitable for large-scale industrial production?
A: Yes, the patent explicitly states the method features mild reaction conditions (0-50°C for key steps) and avoids hazardous reagents like lithium aluminum hydride, making it highly suitable for commercial amplification.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Afuresertib Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from patent literature to commercial reality requires more than just chemical knowledge; it demands engineering excellence and rigorous quality assurance. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the intricate details of the titanium-catalyzed cyanation and subsequent reductions are managed with precision. We understand that the purity specifications for kinase inhibitor intermediates are non-negotiable, which is why our rigorous QC labs employ advanced analytical techniques to monitor enantiomeric excess and trace impurities at every stage of the synthesis, guaranteeing a product that meets the highest global standards.
We invite pharmaceutical companies and research institutions to collaborate with us to optimize their supply chains for Afuresertib and related AKT inhibitors. By leveraging our expertise in this specific patented route, we can help you achieve significant efficiencies and cost reductions. We encourage you to contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your volume requirements. Whether you need specific COA data for the chiral nitrile intermediate or comprehensive route feasibility assessments for the final API, our experts are ready to provide the data-driven insights necessary to accelerate your development timelines.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →