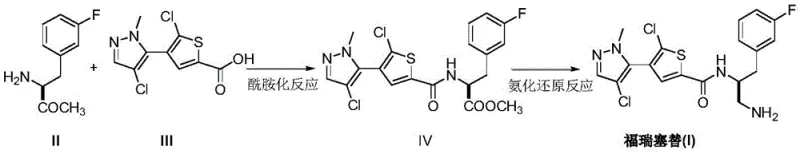
Advanced Manufacturing of Afuresertib via Streamlined Amidation-Reduction Sequence
The pharmaceutical landscape for oncology treatments continues to evolve with the development of potent kinase inhibitors, among which Afuresertib (also known as Forrestitide) stands out as a promising pan-AKT inhibitor. Recent intellectual property developments, specifically patent CN111592531A, have unveiled a transformative preparation method that addresses long-standing synthetic inefficiencies. This patent discloses a novel pathway utilizing an amidation reaction followed by an ammonification reduction reaction between (2S)-2-amino-3-(3-fluorophenyl)methyl propionate and 5-chloro-4-(4-chloro-1-methyl-1H-pyrazol-5-yl)-2-thiophenecarboxylic acid. For R&D directors and procurement specialists alike, this innovation represents a critical pivot point, shifting away from cumbersome multi-step protections toward a more direct, high-yielding convergence strategy that promises to redefine the cost structure and supply reliability of this high-value API intermediate.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art synthesis routes, such as those reported in international patent WO2008098104, rely heavily on complex protection group chemistry to manage the reactivity of the chiral amine center. The traditional synthesis of Intermediate A typically necessitates a sequence involving amino protection, carboxyl reduction, phthalimide ammoniation, and subsequent deprotection. This reliance on phthalimide groups introduces significant operational friction; each protection and deprotection cycle adds at least two distinct unit operations, consuming additional reagents, solvents, and time. Furthermore, the presence of adjacent amino groups in the target structure creates a high risk of competing side reactions, forcing process chemists to adopt these defensive synthetic strategies. The cumulative effect is a lengthened critical path, reduced overall throughput, and a broader impurity profile that complicates downstream purification, ultimately inflating the cost of goods sold (COGS) for the final active pharmaceutical ingredient.
The Novel Approach
In stark contrast, the methodology outlined in CN111592531A bypasses the need for repetitive amino protection by leveraging the inherent stability of the methyl ester functionality during the coupling phase. The core innovation lies in the direct condensation of methyl (2S)-2-amino-3-(3-fluorophenyl)propionate with the functionalized thiophene acid. By avoiding the installation and removal of bulky phthalimide masks, the new route drastically shortens the synthetic timeline. This approach not only simplifies the workflow but also mitigates the formation of byproducts associated with harsh deprotection conditions. The result is a cleaner reaction profile and a more robust process capable of sustaining high purity standards without the burden of excessive chromatographic purification, marking a substantial leap forward in process chemistry efficiency.
Mechanistic Insights into HBTU-Mediated Amidation and Hydride Reduction
The success of this streamlined route hinges on the precise control of the amidation kinetics and the subsequent chemoselective reduction. In the first stage, the coupling of the chiral amino ester and the chloro-thiophene acid is facilitated by advanced peptide coupling reagents such as HBTU (O-(Benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate) or BOP. These uronium salts activate the carboxylic acid to form a highly reactive O-acylisourea or active ester intermediate, which is then rapidly attacked by the nucleophilic amine. The use of strong non-nucleophilic bases like DBU (1,8-Diazabicyclo[5.4.0]undec-7-ene) or DBN ensures rapid deprotonation of the amine salt without inducing racemization at the chiral center, a critical quality attribute for this oncology candidate. Experimental data from the patent indicates that conducting this reaction in polar aprotic solvents like acetonitrile or DMF at moderate temperatures (25-30°C) yields the intermediate amide ester in exceptional quantities, often exceeding 90% isolated yield.
Following the formation of the amide bond, the transformation of the methyl ester into the primary amine requires a powerful yet controlled reduction strategy. The patent details the use of lithium aluminum hydride (LiAlH4) or borane complexes as the reducing agents. This step is mechanistically distinct from simple carbonyl reduction; it involves an initial ammoniation or interaction with the ammonia source followed by hydride delivery to reduce the ester functionality directly to the amine. The choice of solvent is pivotal here, with tetrahydrofuran (THF) serving as the medium for the reduction phase to ensure solubility of the hydride species while maintaining thermal stability. By carefully managing the stoichiometry—using approximately 6 to 12 equivalents of reducing agent—the process ensures complete conversion of the ester while preserving the sensitive chloro-pyrazole and chloro-thiophene moieties, thereby delivering the final Afuresertib molecule with high structural integrity and minimal halogen loss.
How to Synthesize Afuresertib Efficiently
Implementing this novel synthesis route requires strict adherence to the optimized reaction parameters defined in the patent to maximize yield and purity. The process begins with the activation of the thiophene carboxylic acid using coupling agents like HBTU in the presence of a base promoter, followed by the addition of the chiral amino ester. After isolating the intermediate amide ester, the second phase involves a careful ammoniation reduction sequence using lithium aluminum hydride or borane under inert atmosphere. For process engineers and laboratory teams seeking to replicate this high-efficiency pathway, the detailed standardized operating procedures and specific molar ratios are critical for success. Please refer to the structured guide below for the step-by-step technical execution.
- Perform amidation between methyl (2S)-2-amino-3-(3-fluorophenyl)propionate and the thiophene carboxylic acid using HBTU/BOP and DBU/DBN.
- Isolate the intermediate amide ester (IV) through extraction and recrystallization.
- Execute ammoniation reduction using Lithium Aluminum Hydride or Borane to convert the ester to the final primary amine.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this novel synthetic route offers compelling economic and logistical benefits that extend far beyond simple yield improvements. By eliminating the multi-step protection and deprotection sequences associated with phthalimide chemistry, the manufacturing process becomes significantly leaner. This reduction in synthetic steps directly correlates to a decrease in raw material consumption, solvent usage, and waste generation, all of which are primary drivers of manufacturing costs. The simplified workflow also reduces the demand for specialized equipment and labor hours per kilogram of output, allowing for a more competitive pricing structure in the global market for high-purity pharmaceutical intermediates.
- Cost Reduction in Manufacturing: The elimination of expensive protecting group reagents and the associated purification steps leads to a drastic simplification of the bill of materials. Without the need for phthalic anhydride derivatives and harsh deprotection hydrazines, the direct cost of goods is substantially lowered. Furthermore, the high yields reported in the amidation step (over 90%) minimize the loss of valuable chiral starting materials, ensuring that every gram of input contributes effectively to the final output, thereby optimizing the overall cost efficiency of the production campaign.
- Enhanced Supply Chain Reliability: A shorter synthetic route inherently reduces the lead time required for production cycles. With fewer unit operations and intermediate isolations, the turnaround time from raw material intake to finished API intermediate is compressed. This agility allows suppliers to respond more rapidly to fluctuating market demands and urgent clinical trial requirements. Additionally, the use of commercially available and stable reagents like HBTU and standard hydride reducers ensures that the supply chain is not vulnerable to shortages of exotic or highly regulated specialty chemicals, guaranteeing consistent continuity of supply.
- Scalability and Environmental Compliance: The mild reaction conditions, particularly the ambient temperature amidation and controlled reduction phases, make this process highly amenable to scale-up from pilot plant to commercial tonnage. The reduction in solvent volume and the avoidance of hazardous deprotection byproducts align with modern green chemistry principles, simplifying waste treatment and environmental compliance. This environmental optimization not only reduces disposal costs but also future-proofs the manufacturing site against increasingly stringent regulatory standards regarding chemical emissions and effluent management.
Frequently Asked Questions (FAQ)
Understanding the technical nuances of this new preparation method is essential for stakeholders evaluating its adoption for commercial manufacturing. The following questions address common inquiries regarding the reaction mechanism, reagent selection, and scalability potential based on the disclosed patent data. These insights are derived directly from the experimental examples and technical specifications provided in the intellectual property documentation to ensure accuracy and relevance for technical decision-makers.
Q: How does the new route improve upon the conventional phthalimide protection method?
A: The conventional method requires multiple protection and deprotection steps (phthalimide), lengthening the synthesis. The new route utilizes a direct amidation of the amino ester, eliminating these steps and improving overall yield.
Q: What reducing agents are suitable for the final step?
A: The patent specifies Lithium Aluminum Hydride (LiAlH4) or Borane (e.g., Borane dimethylsulfide) as effective reducing agents for converting the intermediate ester to the target amine.
Q: Is this process suitable for industrial scale-up?
A: Yes, the patent explicitly states the method features mild conditions, simple operations, and environment optimization, making it highly suitable for industrial amplification and commercial production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Afuresertib Supplier
As the demand for next-generation AKT inhibitors grows, securing a manufacturing partner with deep technical expertise in complex heterocyclic synthesis is paramount. NINGBO INNO PHARMCHEM stands at the forefront of this capability, leveraging extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our state-of-the-art facilities are equipped to handle the specific requirements of this amidation-reduction sequence, ensuring that stringent purity specifications are met through our rigorous QC labs and advanced analytical instrumentation. We understand that the consistency of the chiral center and the integrity of the halogenated motifs are non-negotiable for clinical efficacy, and our processes are designed to deliver exactly that level of quality assurance.
We invite global pharmaceutical partners to engage with us for a Customized Cost-Saving Analysis tailored to your specific volume requirements. By collaborating with our technical procurement team, you can access specific COA data and comprehensive route feasibility assessments that demonstrate how our optimized synthesis of Afuresertib can enhance your supply chain resilience. Contact us today to discuss how we can support your clinical and commercial needs with reliable, high-quality intermediates produced via this cutting-edge technology.