Scalable Synthesis of Fluorinated Pyrrolizine Intermediates for Next-Gen Oncology Drugs
Scalable Synthesis of Fluorinated Pyrrolizine Intermediates for Next-Gen Oncology Drugs
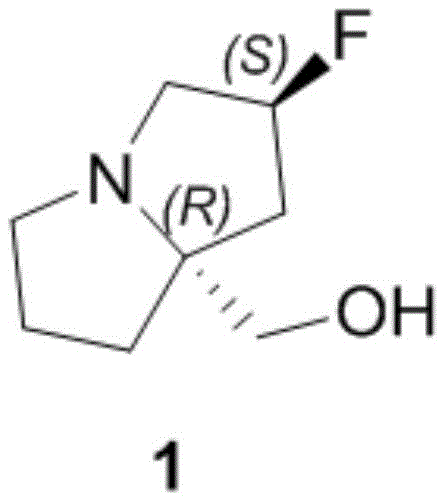
The rapid advancement of targeted oncology therapies has placed immense pressure on the supply chain for complex chiral intermediates, particularly those required for KRAS G12D inhibitors like MRTX1133. A pivotal breakthrough in this domain is detailed in Chinese Patent CN115894503A, which discloses a highly efficient preparation method for the azacyclopentane derivative known as ((2R,7aS)-2-fluorohexahydro-1H-pyrrolazin-7a-yl)methanol. This specific molecular scaffold serves as a critical building block in the design and synthesis of potent anti-tumor agents. The patent introduces a robust synthetic strategy that begins with the abundant and inexpensive chiral pool material, L-proline hydrochloride. By restructuring the synthetic sequence to delay expensive transformations until the final stages, this methodology offers a compelling solution for reliable pharmaceutical intermediate supplier networks seeking to optimize their production pipelines. The structural complexity of the target molecule, featuring a fused bicyclic system with specific fluorine stereochemistry, demands a precise and controlled approach to ensure high optical purity.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to this innovation, the synthesis of this specific pyrrolizine core was fraught with significant operational hazards and economic inefficiencies. Existing literature, such as patent WO2022031678A1, described routes that relied heavily on dangerous and difficult-to-handle reagents. Specifically, the use of ozone for oxidative cleavage and dimethyl sulfide for workup presents severe safety challenges, including explosion risks and the generation of malodorous sulfur waste. Furthermore, these legacy processes often employed DAST (Diethylaminosulfur trifluoride) and lithium aluminum hydride in early or middle stages, which complicates purification and increases the cost of goods sold due to the loss of valuable chiral material if yields are suboptimal. Another reported method in Organic Process Research & Development (2022) offered improvements but still suffered from excessive step counts and cumbersome operational procedures. These factors collectively create bottlenecks for cost reduction in API manufacturing, as the handling of hazardous materials requires specialized equipment and rigorous safety protocols that drive up capital expenditure.
The Novel Approach
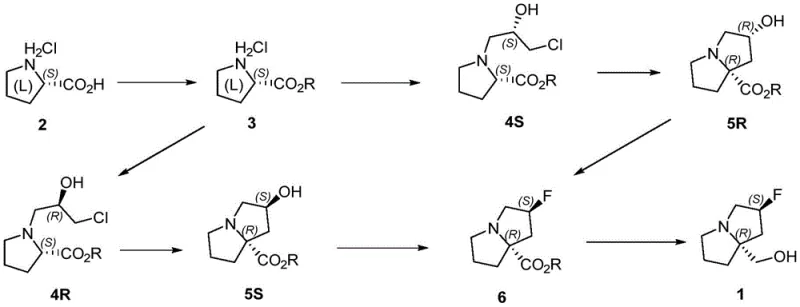
In stark contrast, the methodology outlined in CN115894503A presents a streamlined, five-to-six-step sequence that fundamentally reimagines the construction of the bicyclic core. The strategy capitalizes on the readily available chirality of L-proline hydrochloride, effectively bypassing the need for complex asymmetric catalysis in the initial steps. The route proceeds through a logical progression of esterification, epoxide ring-opening, and a crucial intramolecular cyclization to form the pyrrolizine skeleton. By deferring the high-cost fluorination and reduction steps to the very end of the synthesis, the process minimizes the financial risk associated with processing large quantities of advanced intermediates. This tactical arrangement ensures that the most expensive reagents are utilized only when the molecular complexity is highest, thereby maximizing the value of every gram produced. The result is a pathway that is not only chemically elegant but also commercially viable for the commercial scale-up of complex pharmaceutical intermediates.
Mechanistic Insights into Intramolecular Cyclization and Fluorination
The heart of this synthetic success lies in the precise execution of the intramolecular cyclization step, which transforms the linear chlorohydrin precursor into the rigid bicyclic pyrrolizine system. This transformation typically involves treating the intermediate with a strong, non-nucleophilic base such as lithium hexamethyldisilazide (LiHMDS) or lithium diisopropylamide (LDA) at cryogenic temperatures, often around -78°C. The low temperature is critical for kinetic control, ensuring that the deprotonation occurs selectively at the alpha-position of the ester without triggering unwanted side reactions or racemization. Once the enolate is formed, it undergoes an intramolecular nucleophilic substitution (SN2) attack on the terminal chloride of the side chain. This ring-closing event establishes the quaternary center at the 7a-position with high stereochemical fidelity, locking the relative configuration of the fused rings. The choice of solvent, typically dry tetrahydrofuran or methyl tert-butyl ether, further influences the aggregation state of the lithium enolate, playing a subtle but vital role in determining the diastereomeric ratio of the cyclized product.
Following the construction of the carbon skeleton, the installation of the fluorine atom represents another critical mechanistic challenge. The patent describes the conversion of the secondary hydroxyl group into a fluoride using reagents like DAST or a combination of trifluoromethanesulfonic anhydride and HF-pyridine complexes. This step proceeds via an activation of the hydroxyl group followed by nucleophilic displacement by fluoride. Crucially, this substitution typically occurs with inversion of configuration, which must be accounted for in the design of the precursor stereochemistry to ensure the final product possesses the desired (S)-configuration at the fluorine-bearing carbon. The subsequent reduction of the ester moiety to the primary alcohol, using hydride sources like LiAlH4 or borane, completes the synthesis. Throughout these transformations, impurity control is managed by the high selectivity of the cyclization and the ability to purify intermediates via standard extraction and chromatography techniques before the final steps.
How to Synthesize ((2R,7aS)-2-fluorohexahydro-1H-pyrrolazin-7a-yl)methanol Efficiently
Implementing this synthesis requires strict adherence to the reaction conditions specified in the patent to maintain high yields and optical purity. The process begins with the esterification of L-proline hydrochloride, followed by alkylation with epichlorohydrin under basic conditions. The subsequent cyclization is the most sensitive step, requiring anhydrous conditions and precise temperature control to prevent polymerization or elimination side products. Operators must ensure that the fluorination reagents are handled with appropriate safety measures due to their corrosive nature. For a detailed breakdown of the specific molar ratios, temperatures, and workup procedures for each stage, please refer to the standardized protocol below.
- Esterify L-proline hydrochloride with thionyl chloride in methanol to obtain the methyl ester intermediate.
- React the ester with (S)-epichlorohydrin using a base like DBU or triethylamine to form the chlorohydrin side chain.
- Perform intramolecular cyclization using a strong base such as LiHMDS at low temperature (-78°C) to close the pyrrolizine ring.
- Conduct stereoselective fluorination of the hydroxyl group using DAST or Tf2O/HF-pyridine complexes.
- Reduce the ester functionality to the primary alcohol using LiAlH4 or borane to yield the final target molecule.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement perspective, the shift to this L-proline-based route offers substantial strategic benefits that directly impact the bottom line and supply security. The primary advantage stems from the utilization of L-proline hydrochloride, a commodity chemical produced on a massive scale for the food and pharmaceutical industries. This ensures a stable, multi-source supply of the starting material, effectively insulating the production of this high-value intermediate from the volatility often seen with exotic chiral catalysts or rare starting materials. By anchoring the synthesis in such a robust feedstock, manufacturers can secure long-term contracts at predictable price points, facilitating better budget forecasting for downstream API projects. Furthermore, the elimination of ozone generators and the associated safety infrastructure significantly lowers the barrier to entry for contract manufacturing organizations (CMOs) looking to bid on this chemistry.
- Cost Reduction in Manufacturing: The economic architecture of this route is optimized by pushing the most expensive reagents to the final steps. In traditional synthesis, losing material during a late-stage fluorination is catastrophic; however, by keeping the molecule relatively simple until the end, the overall cost of wasted material is minimized. Additionally, the avoidance of transition metal catalysts removes the need for expensive metal scavenging steps and the rigorous testing required to meet residual metal specifications in the final drug substance. This simplification of the purification train translates directly into lower processing costs and higher throughput, enabling significant cost savings without compromising quality.
- Enhanced Supply Chain Reliability: The reliance on standard organic transformations such as esterification, alkylation, and reduction means that the process can be executed in a wide range of multipurpose chemical facilities. There is no dependency on specialized equipment for high-pressure hydrogenation or cryogenic ozonolysis, which are often bottlenecks in crowded manufacturing schedules. This flexibility allows for rapid technology transfer between sites and ensures that production can be scaled up or shifted geographically with minimal friction. Consequently, lead times for high-purity pharmaceutical intermediates can be drastically reduced, providing a competitive edge in fast-moving oncology drug development programs.
- Scalability and Environmental Compliance: The environmental profile of this new method is markedly superior to prior art. By removing sulfur-based reagents like dimethyl sulfide and avoiding the generation of ozonide byproducts, the waste stream is cleaner and easier to treat. The solvents used, such as methanol, ethyl acetate, and THF, are common and easily recovered through distillation, supporting green chemistry initiatives. This alignment with environmental, social, and governance (ESG) goals is increasingly important for multinational pharma companies selecting suppliers. The simplified waste profile reduces disposal costs and regulatory hurdles, making the commercial scale-up of complex intermediates smoother and more sustainable in the long term.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthetic route. These insights are derived directly from the experimental data and claims within patent CN115894503A, providing clarity on the feasibility and advantages of the technology. Understanding these nuances is essential for technical teams evaluating the route for potential licensing or contract manufacturing partnerships.
Q: What are the critical safety advantages of this new synthetic route compared to prior art?
A: The novel route described in CN115894503A eliminates the use of hazardous reagents such as ozone and dimethyl sulfide, which were required in previous methods (e.g., WO2022031678A1). By utilizing L-proline hydrochloride and standard epoxide chemistry, the process significantly reduces explosion risks and toxic waste generation, making it far more suitable for large-scale industrial production.
Q: How does the stereochemistry control ensure high purity for the KRAS inhibitor intermediate?
A: The process leverages the inherent chirality of L-proline hydrochloride as the starting material. Through a carefully controlled intramolecular cyclization step using bases like LiHMDS at -78°C, the stereochemical integrity is maintained throughout the ring-closing sequence. This ensures the formation of the specific (2R,7aS) configuration required for biological activity in downstream KRAS G12D inhibitors.
Q: Is this synthesis route scalable for commercial API manufacturing?
A: Yes, the route is specifically designed for scalability. It relies on cheap, commodity chemicals like L-proline and epichlorohydrin rather than exotic catalysts. Furthermore, by pushing high-cost steps like fluorination and reduction to the end of the sequence, the overall economic efficiency is maximized, allowing for cost-effective production from kilogram to multi-ton scales.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable ((2R,7aS)-2-fluorohexahydro-1H-pyrrolazin-7a-yl)methanol Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful development of next-generation KRAS inhibitors depends on the availability of high-quality, stereochemically pure intermediates. Our technical team has thoroughly analyzed the route disclosed in CN115894503A and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. We understand the critical nuances of cryogenic cyclization and safe fluorination chemistry, ensuring that every batch meets stringent purity specifications. Our rigorous QC labs are equipped to verify the optical purity and impurity profile of this complex bicyclic scaffold, guaranteeing that the material performs consistently in your downstream coupling reactions.
We invite you to discuss how our manufacturing capabilities can support your pipeline. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data from pilot batches and comprehensive route feasibility assessments to demonstrate how we can become your trusted partner in bringing life-saving oncology therapies to market faster and more efficiently.