Optimizing Brigatinib Production: A Technical Analysis of High-Yield Synthetic Routes for Commercial Scale-Up
Introduction to Advanced Brigatinib Synthesis Technology
The pharmaceutical landscape for oncology treatments demands not only potent active ingredients but also robust, scalable, and cost-effective manufacturing processes. Patent CN111138492B introduces a significant technological breakthrough in the preparation of Brigatinib (AP26113), a second-generation ALK inhibitor used for treating non-small cell lung cancer. This proprietary methodology addresses critical bottlenecks found in earlier synthetic routes, specifically targeting low yields and complex purification challenges that have historically hindered mass production. By leveraging optimized nucleophilic substitution reactions and efficient catalytic hydrogenation, this new protocol achieves a final product purity exceeding 99.9% with an overall yield surpassing 50%. For global procurement teams and R&D directors, understanding the nuances of this improved synthesis is vital for securing a reliable pharmaceutical intermediates supplier capable of meeting the rigorous demands of the generic drug market.
The strategic value of this patent lies in its ability to streamline the supply chain for high-purity oncology APIs. Traditional methods often suffer from yield losses at every stage, compounding costs and extending lead times. In contrast, the approach detailed in CN111138492B utilizes a convergent strategy where two key intermediates are synthesized independently and then coupled. This modularity allows for better quality control at each stage, ensuring that impurities are managed before the final coupling step. Furthermore, the elimination of column chromatography in favor of crystallization and filtration represents a paradigm shift towards greener, more economical chemistry. As we delve deeper into the technical specifics, it becomes clear that this process offers a viable pathway for cost reduction in pharmaceutical manufacturing while maintaining the highest standards of chemical integrity.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Brigatinib has been plagued by inefficiencies that render large-scale production economically unviable. The original route developed by Ariad Pharmaceuticals, as disclosed in patent WO2016065028(A1), while chemically straightforward, suffers from a dismal final product yield of less than 25%. This low efficiency is primarily due to the reliance on column chromatography for purification at multiple stages, a technique that is notoriously difficult to scale and expensive to operate in an industrial setting. Additionally, alternative routes such as those described in WO2017016410A1 and WO2009070740(A2) introduce their own set of complications, including the use of unstable and toxic reagents like cyanamide, or expensive starting materials that erode profit margins in a competitive generic market.
Beyond economic concerns, the impurity profile of conventional methods poses a significant regulatory risk. Processes described in prior art often generate structurally similar byproducts, such as Impurity 17 in the Suzhou Miracpharma route, which are difficult to separate and can persist in the final API. The presence of such impurities necessitates rigorous and costly analytical testing and additional purification steps, further delaying time-to-market. Moreover, the use of harsh reaction conditions in some legacy routes can lead to the degradation of sensitive functional groups, resulting in lower batch consistency. For a supply chain head, these variables translate into unpredictable delivery schedules and potential supply disruptions, highlighting the urgent need for a more robust and reliable manufacturing protocol.
The Novel Approach
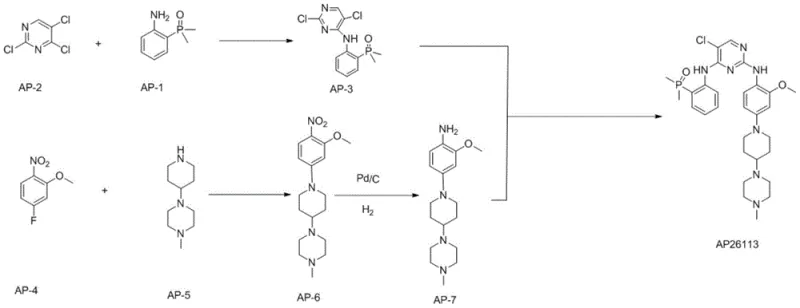
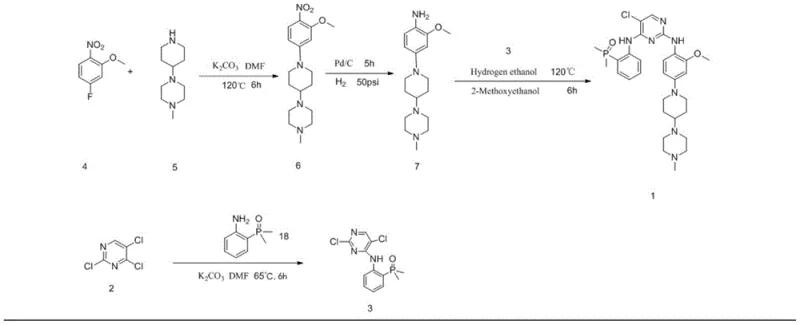
The methodology presented in CN111138492B offers a transformative solution to these longstanding challenges by re-engineering the synthetic pathway for maximum efficiency and purity. This novel approach divides the synthesis into two parallel streams that converge in the final step, allowing for the independent optimization of each fragment. The first stream involves the substitution of 2,4,5-trichloropyrimidine with 2-(dimethylphosphoryl)aniline to form Intermediate AP-3, while the second stream constructs the piperazine-piperidine scaffold through a sequence of substitution and reduction reactions to yield Intermediate AP-7. By isolating these pathways, manufacturers can ensure that each intermediate meets strict purity specifications before the final coupling, thereby minimizing the formation of complex side products.
A defining feature of this new route is its reliance on scalable unit operations rather than laboratory-scale techniques. The replacement of column chromatography with simple workup procedures such as acid-base extraction, slurry washing, and recrystallization dramatically lowers the barrier to commercial scale-up. For instance, the final purification of Brigatinib is achieved through recrystallization from methanol, a solvent that is inexpensive and easy to recover. This shift not only reduces the direct cost of goods sold but also simplifies the environmental footprint of the manufacturing process by reducing solvent waste. Consequently, this approach positions manufacturers as a reliable pharmaceutical intermediates supplier capable of delivering high volumes of API without the logistical nightmares associated with traditional purification methods.
Mechanistic Insights into Nucleophilic Substitution and Catalytic Hydrogenation
The chemical elegance of this synthesis lies in the precise control of reaction mechanisms to favor the desired product while suppressing side reactions. In the formation of Intermediate AP-3, the reaction between 2,4,5-trichloropyrimidine and 2-(dimethylphosphoryl)aniline is a classic nucleophilic aromatic substitution. The use of a mild inorganic base like dipotassium hydrogen phosphate (K2HPO4) in DMF at 65°C is critical; it provides sufficient basicity to deprotonate the aniline without promoting the hydrolysis of the chloro-pyrimidine ring or the phosphoryl group. This careful selection of reaction conditions ensures that the substitution occurs selectively at the 4-position of the pyrimidine ring, preserving the chlorine atoms necessary for the subsequent coupling step. The mechanism avoids the formation of bis-substituted byproducts, which are common when stronger bases or higher temperatures are employed.
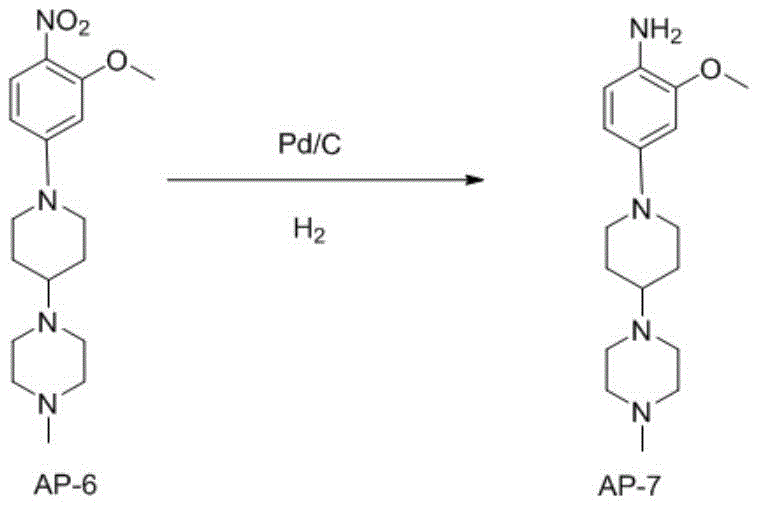
Similarly, the reduction of the nitro group in Intermediate AP-6 to form the aniline Intermediate AP-7 demonstrates the power of catalytic hydrogenation in process chemistry. Using palladium on carbon (Pd/C) in ethanol under mild hydrogen pressure (5-20 psi) allows for the clean conversion of the nitro group to an amine without affecting other sensitive functionalities, such as the methoxy group or the piperazine rings. This step is crucial because alternative reduction methods, such as metal-acid reductions, often generate heavy metal waste and require complex workups. The catalytic method described here is atom-economical and generates water as the only byproduct. Furthermore, the process effectively avoids the formation of Impurity B, a reduced byproduct observed in other routes, by strictly controlling the reaction time and catalyst loading, ensuring a high-purity amine ready for the final condensation.
How to Synthesize Brigatinib Efficiently
Implementing this optimized synthesis requires a disciplined approach to reaction parameters and workup procedures to fully realize the benefits of yield and purity. The process is designed to be robust, tolerating slight variations in stoichiometry while maintaining high performance, which is essential for industrial reproducibility. Key to the success of this route is the management of the acid-binding agents and solvents in the initial substitution steps, as well as the precise control of temperature during the final coupling reaction. By adhering to the specific conditions outlined in the patent, manufacturers can consistently achieve yields above 90% for individual steps, culminating in a highly efficient overall process. The following guide outlines the standardized operational framework derived from the patent examples, serving as a blueprint for technical teams aiming to adopt this superior methodology.
- React 2,4,5-trichloropyrimidine with 2-(dimethylphosphoryl)aniline using a base like K2HPO4 in DMF to form Intermediate AP-3.
- Perform nucleophilic substitution between 2-nitro-5-fluoroanisole and 1-methyl-4-(4-piperidinyl)piperazine in acetonitrile to yield Intermediate AP-6.
- Reduce the nitro group of Intermediate AP-6 to an amine using Pd/C catalytic hydrogenation in ethanol to obtain Intermediate AP-7.
- Condense Intermediate AP-3 and Intermediate AP-7 under acidic catalysis in dimethoxyethanol, followed by recrystallization to purify Brigatinib.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain executives, the adoption of the CN111138492B synthesis route translates into tangible strategic advantages that extend beyond mere chemical yield. The primary driver of value is the substantial cost reduction in pharmaceutical manufacturing achieved by eliminating expensive and inefficient processing steps. The removal of column chromatography, which is a major cost center in API production due to high solvent consumption and silica gel usage, directly improves the gross margin of the final product. Additionally, the use of commercially available and inexpensive starting materials, such as 2,4,5-trichloropyrimidine and 2-nitro-5-fluoroanisole, ensures that raw material costs remain stable and predictable, shielding the supply chain from volatility associated with exotic reagents.
- Cost Reduction in Manufacturing: The economic impact of this process is profound, driven by the simplification of downstream processing. By replacing column chromatography with crystallization and filtration, the process significantly reduces solvent usage and waste disposal costs, which are often hidden expenses in traditional API synthesis. The high yield of each step, particularly the 90.3% yield in the formation of Intermediate AP-3 and the 95% yield in the hydrogenation step, means that less raw material is wasted, further driving down the cost per kilogram. This efficiency allows for a more competitive pricing structure in the generic market, where margin compression is a constant challenge.
- Enhanced Supply Chain Reliability: From a logistics perspective, the robustness of this synthetic route enhances supply security. The reliance on stable, non-hazardous reagents and standard solvents like ethanol and acetonitrile minimizes the risk of supply disruptions caused by regulatory restrictions on hazardous chemicals. Furthermore, the short reaction times and simple workups reduce the overall cycle time for production batches, enabling manufacturers to respond more quickly to fluctuations in market demand. This agility is critical for maintaining continuous supply to downstream formulation partners and avoiding stockouts that could impact patient access to medication.
- Scalability and Environmental Compliance: The design of this process inherently supports commercial scale-up of complex pharmaceutical intermediates. The unit operations employed—stirred tank reactions, filtration, and distillation—are standard in modern chemical plants, requiring no specialized equipment. This ease of transfer from lab to plant accelerates the timeline for technology transfer and validation. Moreover, the reduced solvent intensity and absence of heavy metal catalysts (other than recoverable Pd/C) align with increasingly stringent environmental regulations, reducing the compliance burden and enhancing the sustainability profile of the manufacturing operation.
Frequently Asked Questions (FAQ)
To further clarify the technical and commercial implications of this synthesis method, we have compiled answers to common inquiries regarding its implementation and performance. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering a transparent view of the technology's capabilities. Understanding these details is essential for stakeholders evaluating the feasibility of integrating this route into their existing production portfolios or sourcing strategies.
Q: How does this new synthesis route improve upon the original Ariad process?
A: The new route described in CN111138492B significantly improves the overall yield to over 50% compared to less than 25% in the original Ariad process. Crucially, it eliminates the need for column chromatography, replacing it with simple filtration and recrystallization, which drastically reduces production costs and facilitates industrial scale-up.
Q: What specific impurities are controlled in this manufacturing process?
A: The process specifically addresses and avoids the formation of Impurity A, Impurity B, and Impurity C, which are common byproducts in conventional routes. By optimizing reaction conditions such as temperature and acid-binding agents, the method ensures a final product purity exceeding 99.9%, meeting stringent regulatory requirements for oncology medications.
Q: Is this synthesis method suitable for large-scale commercial production?
A: Yes, the method is designed for scalability. It utilizes readily available raw materials and avoids expensive reagents and complex purification steps like column chromatography. The use of standard unit operations such as slurry, extraction, and crystallization makes it highly adaptable for multi-ton annual production capacities.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Brigatinib Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from patent to production requires a partner with deep technical expertise and proven manufacturing capabilities. Our team has extensively analyzed the CN111138492B process and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. We are equipped with state-of-the-art facilities capable of handling the specific solvent systems and reaction conditions required for Brigatinib synthesis, ensuring that every batch meets stringent purity specifications. Our rigorous QC labs employ advanced analytical techniques to monitor impurity profiles, guaranteeing that the final API complies with global pharmacopoeia standards and is free from the critical impurities identified in legacy routes.
We invite potential partners to engage with our technical procurement team to discuss how this optimized synthesis can benefit your supply chain. By leveraging our expertise, you can secure a consistent supply of high-quality Brigatinib while achieving significant operational efficiencies. We encourage you to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. Our team is ready to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing prowess can support your long-term strategic goals in the oncology therapeutic area.