Advanced Synthetic Route for Repaglinide Intermediates: Scaling High-Purity Diabetes Medication Production
Advanced Synthetic Route for Repaglinide Intermediates: Scaling High-Purity Diabetes Medication Production
The global demand for effective type II diabetes treatments continues to surge, placing immense pressure on pharmaceutical supply chains to deliver high-purity active pharmaceutical ingredients (APIs) with consistent quality and cost-efficiency. In this landscape, the synthetic methodology disclosed in Chinese Patent CN101481363B represents a significant technological leap forward for the manufacturing of repaglinide, a potent meglitinide analog. This patent details a refined preparation method that addresses critical bottlenecks found in earlier generations of synthesis, specifically focusing on the optimization of the amidation step and the subsequent deprotection sequences. By introducing a specific protecting group strategy and utilizing safer condensing agents, this route offers a compelling value proposition for manufacturers seeking to enhance their production capabilities. For procurement leaders and R&D directors alike, understanding the nuances of this improved pathway is essential for securing a reliable repaglinide intermediate supplier capable of meeting rigorous international standards.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial synthesis of repaglinide has relied heavily on routes described in foundational patents such as US5312924, which typically involve the direct condensation of a free amine with a carboxylic acid derivative. As illustrated in the reaction scheme below, these conventional pathways often necessitate the use of harsh condensing agents like dicyclohexylcarbodiimide (DCC) or toxic combinations involving triphenylphosphine and carbon tetrachloride. 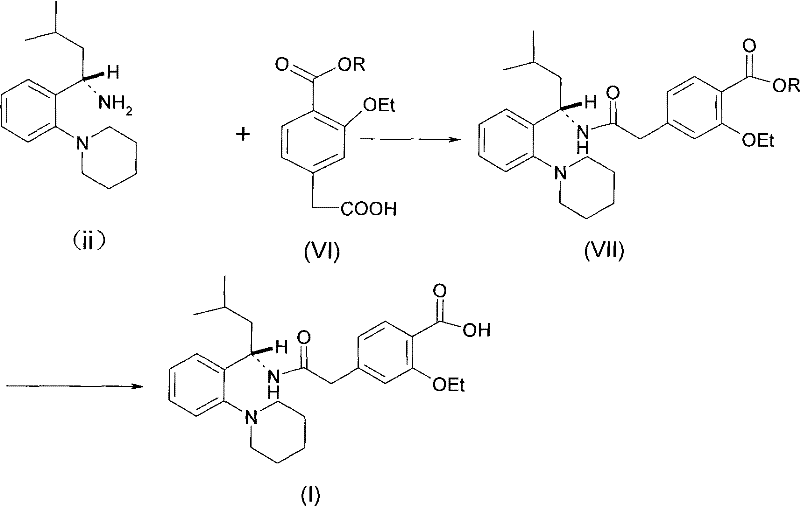 The reliance on such reagents introduces severe operational challenges, including the generation of tenacious byproducts like dicyclohexylurea, which are notoriously difficult to remove completely from the final API, thereby compromising purity profiles. Furthermore, these traditional methods are frequently plagued by extended reaction times and lower overall yields, which directly inflate manufacturing costs and extend lead times for high-purity pharmaceutical intermediates. The environmental and safety liabilities associated with handling large quantities of toxic solvents and reagents also pose significant compliance risks for modern chemical facilities aiming for green chemistry certification.
The reliance on such reagents introduces severe operational challenges, including the generation of tenacious byproducts like dicyclohexylurea, which are notoriously difficult to remove completely from the final API, thereby compromising purity profiles. Furthermore, these traditional methods are frequently plagued by extended reaction times and lower overall yields, which directly inflate manufacturing costs and extend lead times for high-purity pharmaceutical intermediates. The environmental and safety liabilities associated with handling large quantities of toxic solvents and reagents also pose significant compliance risks for modern chemical facilities aiming for green chemistry certification.
The Novel Approach
In stark contrast to the legacy methods, the innovative process outlined in CN101481363B employs a strategic protection-deprotection manifold that fundamentally alters the reaction kinetics and thermodynamics. The core of this advancement lies in the utilization of a protected amine intermediate, specifically the (S)-type compound shown in Formula II, which reacts with the acid component (Formula III) under much milder conditions. 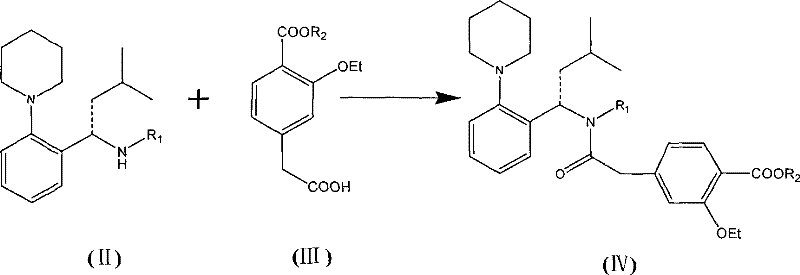 By selecting carbonyldiimidazole (CDI) as the preferred condensing agent, the new route eliminates the formation of insoluble urea byproducts, thereby streamlining the downstream purification process and drastically reducing solvent consumption. This approach not only shortens the reaction time significantly but also enhances the safety profile of the operation by removing the need for carcinogenic solvents like carbon tetrachloride. For a cost reduction in API manufacturing, this shift translates to fewer processing steps, lower waste disposal costs, and a more robust process that is inherently easier to scale from pilot plant to commercial tonnage without sacrificing quality.
By selecting carbonyldiimidazole (CDI) as the preferred condensing agent, the new route eliminates the formation of insoluble urea byproducts, thereby streamlining the downstream purification process and drastically reducing solvent consumption. This approach not only shortens the reaction time significantly but also enhances the safety profile of the operation by removing the need for carcinogenic solvents like carbon tetrachloride. For a cost reduction in API manufacturing, this shift translates to fewer processing steps, lower waste disposal costs, and a more robust process that is inherently easier to scale from pilot plant to commercial tonnage without sacrificing quality.
Mechanistic Insights into CDI-Mediated Amidation and Sequential Deprotection
The mechanistic elegance of this synthesis is rooted in the electronic modulation of the amine nucleophile through the introduction of the R1 radical, preferably a p-methoxybenzyl group. In traditional free-amine couplings, steric hindrance around the chiral center can often retard the attack on the activated carboxyl species, leading to incomplete conversion and racemization risks. However, the electron-donating nature of the p-methoxybenzyl group increases the electron density on the nitrogen atom, thereby boosting its nucleophilicity and facilitating a rapid and selective attack on the acyl-imidazole intermediate formed by CDI activation.  This enhancement in reactivity allows the reaction to proceed efficiently at room temperature, preserving the stereochemical integrity of the chiral center which is critical for the biological activity of the final drug. Furthermore, the use of CDI generates imidazole as a byproduct, which is highly soluble in aqueous workups, ensuring that the crude product stream is significantly cleaner than those generated by DCC-mediated processes, thus simplifying the path to high-purity OLED material or pharmaceutical grade standards.
This enhancement in reactivity allows the reaction to proceed efficiently at room temperature, preserving the stereochemical integrity of the chiral center which is critical for the biological activity of the final drug. Furthermore, the use of CDI generates imidazole as a byproduct, which is highly soluble in aqueous workups, ensuring that the crude product stream is significantly cleaner than those generated by DCC-mediated processes, thus simplifying the path to high-purity OLED material or pharmaceutical grade standards.
Following the amidation, the deprotection strategy employs a sophisticated sequential hydrolysis protocol that maximizes yield and minimizes degradation. The process first targets the ester protecting group (R2) using a mild inorganic base such as sodium hydroxide or potassium carbonate, which selectively cleaves the ester bond without disturbing the sensitive amide linkage or the benzyl-type amine protection. Subsequently, the robust amine protecting group (R1) is removed under acidic conditions using a mixed acid system of trifluoroacetic acid and methanesulfonic acid. This two-step deprotection is superior to simultaneous hydrolysis methods, which often require harsher conditions that can lead to epimerization or decomposition of the sensitive benzoic acid moiety. By controlling the pH and reagent stoichiometry precisely, manufacturers can ensure that the final repaglinide product meets stringent purity specifications with minimal impurity carryover.
How to Synthesize Repaglinide Efficiently
The practical implementation of this patented methodology requires careful attention to reagent quality and process parameters to fully realize its benefits in a commercial setting. The synthesis begins with the preparation of the key chiral amine intermediate, which can be derived from commercially available ketones through dehydration, reduction, and resolution, ensuring a consistent supply of the correct enantiomer. Once the protected amine is secured, the amidation with the ethoxy-benzoic acid derivative is conducted in a polar aprotic solvent like acetonitrile, where the activation by CDI proceeds rapidly to form the protected amide precursor. The detailed standardized synthesis steps, including specific molar ratios, temperature controls, and workup procedures required to achieve optimal results, are outlined in the guide below.
- Condense the (S)-type protected amine (Formula II) with the ethoxy-benzoic acid derivative (Formula III) using CDI as a condensing agent to form the protected amide (Formula IV).
- Perform alkaline hydrolysis on Formula IV using sodium hydroxide or potassium hydroxide to remove the ester protecting group (R2).
- Execute acid hydrolysis using a mixed acid system of trifluoroacetic acid and methanesulfonic acid to remove the amine protecting group (R1), yielding pure repaglinide.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this novel synthetic route offers tangible strategic advantages that extend beyond simple chemical yield improvements. The elimination of hazardous reagents and the simplification of purification steps directly correlate to a reduction in operational expenditure (OPEX) and a decrease in the environmental footprint of the manufacturing site. By switching to a process that utilizes safer, more common reagents like CDI and sodium hydroxide, facilities can reduce their dependency on specialized waste treatment protocols and lower their insurance premiums related to chemical handling risks. This transition supports a more resilient supply chain by mitigating the risk of production stoppages due to regulatory scrutiny or reagent shortages, ensuring a steady flow of critical diabetes medication intermediates to the market.
- Cost Reduction in Manufacturing: The replacement of expensive and problematic condensing agents like DCC with CDI, combined with the avoidance of toxic solvents, leads to substantial cost savings in raw material procurement and waste management. The cleaner reaction profile reduces the need for extensive chromatographic purification, allowing for more efficient crystallization-based isolation which is far more economical at scale. Additionally, the shorter reaction times increase reactor throughput, enabling the same equipment to produce significantly more batches per year, effectively lowering the fixed cost allocation per kilogram of product.
- Enhanced Supply Chain Reliability: The reliance on commercially available starting materials, such as the acid component (Formula III) and standard protecting group reagents, ensures that the supply chain is not vulnerable to single-source bottlenecks. The robustness of the reaction conditions, which tolerate minor variations in temperature and stoichiometry better than the sensitive legacy routes, means that production schedules are more predictable and less prone to batch failures. This reliability is crucial for maintaining just-in-time inventory levels and meeting the demanding delivery windows of global pharmaceutical partners.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, utilizing unit operations that are standard in the fine chemical industry, such as liquid-liquid extraction and crystallization, rather than complex or hazardous transformations. The absence of heavy metal catalysts and carcinogenic solvents aligns perfectly with modern environmental, health, and safety (EHS) regulations, facilitating easier permitting and audit compliance. This 'green' advantage not only future-proofs the manufacturing asset but also appeals to end-clients who are increasingly prioritizing sustainability in their vendor selection criteria.
Frequently Asked Questions (FAQ)
To further clarify the technical and commercial implications of this advanced synthesis method, we have compiled a set of frequently asked questions based on the specific details found in the patent literature. These insights address common concerns regarding impurity profiles, scalability, and the specific advantages of the protecting group strategy employed. Understanding these nuances is vital for technical teams evaluating the feasibility of integrating this route into their existing manufacturing portfolios or for sourcing partners who need to validate the quality of the intermediates supplied.
Q: What are the advantages of using CDI over DCC in repaglinide synthesis?
A: According to patent CN101481363B, using Carbonyldiimidazole (CDI) avoids the formation of difficult-to-remove dicyclohexylurea byproducts associated with DCC, significantly simplifying purification and reducing toxicity risks in large-scale manufacturing.
Q: How does the R1 protecting group improve the reaction yield?
A: The introduction of the p-methoxybenzyl group (R1) enhances the nucleophilicity of the amino group, facilitating a faster and more complete condensation reaction while preventing unwanted side reactions on the nitrogen atom.
Q: Is this process suitable for multi-ton commercial production?
A: Yes, the process is designed for industrialization by utilizing commercially available starting materials, avoiding hazardous reagents like carbon tetrachloride, and employing a robust stepwise deprotection strategy that ensures high purity and safety.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Repaglinide Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition to a superior synthetic route requires a partner with deep technical expertise and proven execution capabilities. As a leading CDMO, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of patent CN101481363B are fully realized in practice. Our state-of-the-art facilities are equipped with rigorous QC labs and advanced analytical instrumentation to guarantee that every batch of repaglinide intermediate meets stringent purity specifications, providing our clients with the confidence needed to navigate complex regulatory filings.
We invite you to collaborate with us to leverage this innovative technology for your next project. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements, demonstrating exactly how this route can optimize your bottom line. Please contact us today to request specific COA data and route feasibility assessments, and let us help you secure a competitive edge in the global diabetes care market through superior chemical manufacturing.