Revolutionizing Anticancer Intermediate Production with Safer High-Yield Synthesis Routes
Introduction to Advanced Tricyclic Derivative Synthesis
The pharmaceutical industry continuously seeks robust manufacturing pathways for potent anticancer agents, particularly those mimicking the efficacy of colchicine or paclitaxel with reduced toxicity profiles. Patent CN102548958A introduces a groundbreaking methodology for preparing tricyclic derivatives and their critical intermediates with exceptional yield and purity. This technology addresses the longstanding challenges of industrial scalability by replacing hazardous reagents and inefficient purification techniques with safer, cost-effective alternatives. The core innovation lies in a multi-step synthesis that avoids the use of highly reactive lithium borohydride and eliminates the need for column chromatography, thereby ensuring high productivity and economic feasibility. For global procurement teams, this represents a shift towards more reliable supply chains for complex pharmaceutical intermediates, enabling the consistent production of high-quality active pharmaceutical ingredients (APIs) required for next-generation oncology treatments.
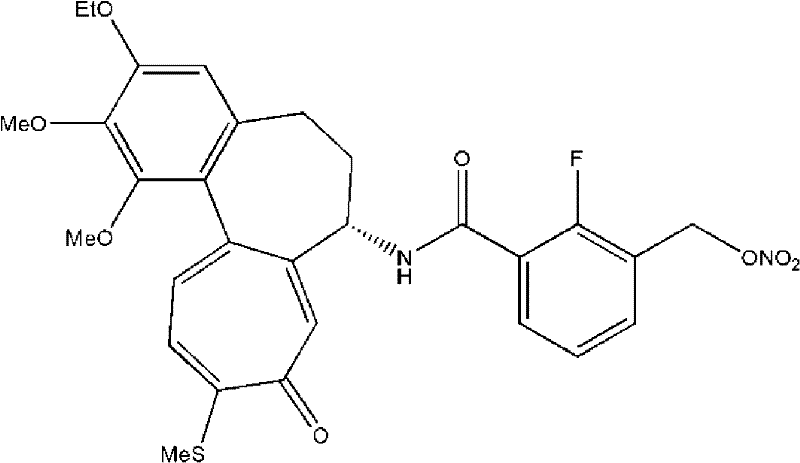
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of key intermediates like 2-fluoro-3-(hydroxymethyl)benzoic acid has been plagued by significant safety and efficiency bottlenecks. As illustrated in Reaction Scheme 4, traditional routes often rely on lithium borohydride (LiBH4) as a reducing agent. This reagent is notoriously unstable, reacting violently with moisture to generate flammable hydrogen gas, creating an unacceptable fire hazard for large-scale manufacturing facilities. Furthermore, conventional purification strategies depend heavily on silica gel column chromatography. While effective for gram-scale laboratory synthesis, this technique is economically prohibitive for industrial production due to the massive consumption of expensive silica and the generation of substantial chemical waste. Additionally, alternative low-temperature lithiation methods involving sec-butyllithium at -78°C present operational complexities and safety risks that hinder commercial adoption.
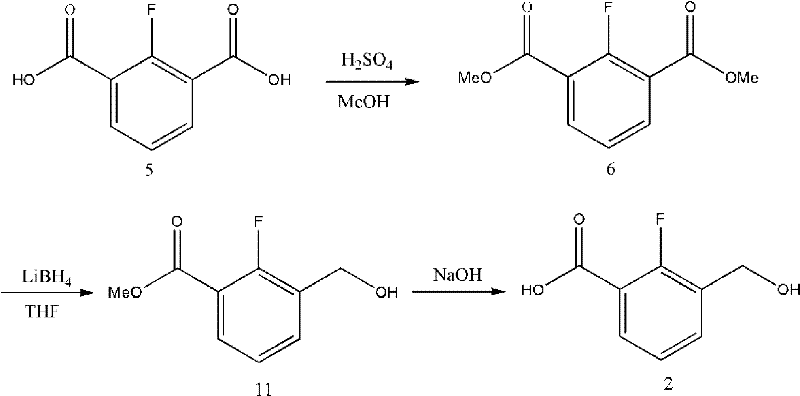
The Novel Approach
The patented methodology fundamentally reengineers the synthetic pathway to prioritize safety and scalability without compromising chemical integrity. By substituting lithium borohydride with sodium borohydride (NaBH4) or lithium aluminum hydride (LiAlH4), the process drastically reduces fire risks while maintaining high reduction efficiency. A pivotal advancement is the strategic introduction of an amide bond prior to the reduction step. This structural modification acts as a protecting group, preventing the over-reduction of ester groups that typically plagues direct reduction attempts. Consequently, the process achieves superior selectivity. Moreover, the replacement of column chromatography with optimized recrystallization protocols—utilizing solvent systems like acetonitrile and ethyl acetate—ensures that impurities are removed efficiently. This shift not only enhances the purity of the final tricyclic derivative to levels exceeding 99% but also aligns with green chemistry principles by minimizing solid waste generation.
Mechanistic Insights into Selective Reduction and Amidation
The chemical elegance of this process lies in its precise control over functional group reactivity. The synthesis begins with the esterification of 2-fluoroisophthalic acid, followed by a controlled hydrolysis to generate a mono-ester. The critical mechanistic step involves the formation of an amide bond using a cyclic amine such as piperidine. This amidation is not merely a structural change; it electronically deactivates the adjacent carbonyl system, allowing for the selective reduction of the remaining ester group to a primary alcohol using sodium borohydride. In the absence of this amide protection, standard reducing agents would indiscriminately reduce both ester functionalities, leading to a diol byproduct and destroying the synthetic utility of the molecule. Following reduction, the amide bond is hydrolyzed under basic conditions to regenerate the carboxylic acid, yielding the desired intermediate with high fidelity. This sequence demonstrates a sophisticated understanding of chemoselectivity, ensuring that the complex molecular architecture required for biological activity is preserved throughout the synthesis.
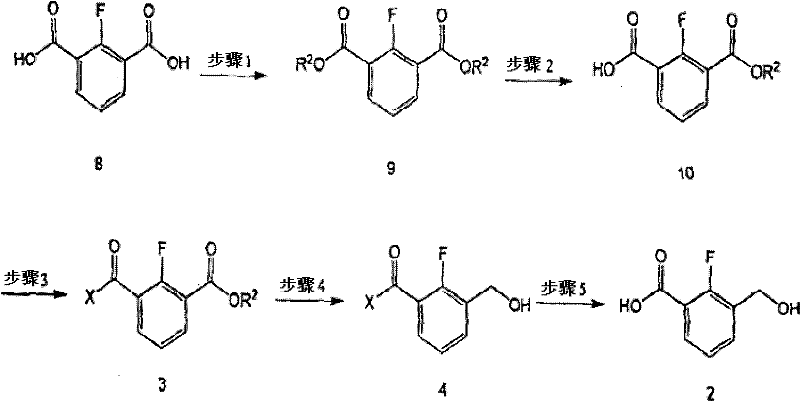
Impurity control is rigorously managed through the physical properties of the intermediates. The patent specifies that recrystallization temperatures between 0°C and 25°C are optimal for purifying the coupling compounds and sulfonated intermediates. By leveraging the differential solubility of the target molecule versus side products in specific solvent mixtures, the process effectively excludes structurally similar impurities that might arise from incomplete reactions or over-reduction. This physical purification method is far more robust than chromatographic separation for bulk manufacturing, as it provides a consistent crystal lattice that rejects impurities during growth. The result is a product with a defined polymorphic form and consistent purity profile, which is essential for regulatory compliance in pharmaceutical manufacturing. This level of control ensures that downstream coupling reactions proceed with predictable kinetics, minimizing the risk of batch failures in the final API synthesis.
How to Synthesize 2-Fluoro-3-(hydroxymethyl)benzoic Acid Efficiently
The standardized protocol for generating this critical building block involves a five-step sequence designed for maximum throughput and safety. The process initiates with the acid-catalyzed esterification of 2-fluoroisophthalic acid in methanol, followed by selective partial hydrolysis using potassium hydroxide to isolate the mono-methyl ester. Subsequent activation of the carboxylic acid via a mixed anhydride method allows for efficient coupling with piperidine. The resulting amide-ester is then subjected to reduction with sodium borohydride in tetrahydrofuran, selectively converting the ester to a hydroxymethyl group. Finally, basic hydrolysis cleaves the amide to release the free acid. Detailed standard operating procedures for each reaction condition, including specific molar ratios and temperature controls, are outlined in the technical guide below to ensure reproducibility across different manufacturing sites.
- Esterify 2-fluoroisophthalic acid with methanol using sulfuric acid catalyst to form dimethyl 2-fluoroisophthalate.
- Perform selective hydrolysis using potassium hydroxide to obtain the mono-ester 2-fluoro-3-(methoxycarbonyl)benzoic acid.
- React the mono-ester with piperidine via mixed anhydride method to form the amide intermediate.
- Reduce the ester group selectively using sodium borohydride to introduce the hydroxymethyl group.
- Hydrolyze the amide bond under basic conditions to yield the final 2-fluoro-3-(hydroxymethyl)benzoic acid.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the transition to this patented synthesis route offers profound strategic benefits beyond mere technical superiority. The elimination of hazardous reagents like lithium borohydride and sec-butyllithium significantly lowers the operational risk profile of the manufacturing facility, reducing insurance costs and the need for specialized containment infrastructure. Furthermore, the shift from column chromatography to recrystallization represents a massive reduction in consumable costs, as the expense of industrial-grade silica gel and the associated waste disposal fees are completely removed from the cost of goods sold (COGS). This process optimization translates directly into improved margin potential for the final API, making it a highly attractive candidate for generic drug development or cost-sensitive therapeutic areas.
- Cost Reduction in Manufacturing: The replacement of expensive and wasteful column chromatography with simple recrystallization techniques leads to substantial cost savings in raw materials and waste management. By avoiding the use of tons of silica gel per kilogram of product, the process dramatically lowers the variable costs associated with purification. Additionally, the higher yields achieved in the intermediate steps (improving from roughly 40% to 75% in key stages) mean that less starting material is required to produce the same amount of final product, further driving down the unit cost of the pharmaceutical intermediate.
- Enhanced Supply Chain Reliability: The use of common, stable reagents like sodium borohydride and potassium hydroxide ensures that the supply chain is not vulnerable to the shortages or price volatility often associated with specialized organometallic reagents. The robustness of the recrystallization purification method also means that production is less susceptible to variations in raw material quality, ensuring consistent output even when sourcing from multiple suppliers. This stability is crucial for maintaining continuous production schedules and meeting the rigorous delivery timelines demanded by global pharmaceutical partners.
- Scalability and Environmental Compliance: The process is inherently designed for scale-up, utilizing standard unit operations like reflux, filtration, and crystallization that are easily implemented in multi-ton reactors. The reduction in hazardous waste generation aligns with increasingly stringent environmental regulations, simplifying the permitting process for new manufacturing lines. By minimizing the use of toxic solvents and eliminating heavy metal or pyrophoric reagent residues, the facility can maintain a cleaner environmental footprint, which is increasingly a prerequisite for partnerships with major multinational pharmaceutical corporations focused on sustainability.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, offering clarity on safety, purity, and scalability. Understanding these factors is essential for technical teams evaluating the feasibility of adopting this route for commercial production. The answers highlight the specific advantages over prior art methods, providing a clear rationale for the technology's superiority in an industrial setting.
Q: Why is the new synthesis method safer than conventional lithium borohydride routes?
A: Conventional methods utilize lithium borohydride (LiBH4), which reacts violently with water to produce flammable gases and poses a severe fire risk. The patented method replaces this with sodium borohydride (NaBH4) or lithium aluminum hydride (LiAlH4), which have significantly lower fire risks and are manageable for industrial mass production.
Q: How does this process improve purity compared to column chromatography?
A: Traditional purification relies on column chromatography, which is economically unfeasible for large scales due to excessive silica gel usage and waste generation. This invention utilizes recrystallization (e.g., using acetonitrile/ethyl acetate mixtures), which achieves high purity (up to 99.8%) while eliminating the cost and environmental burden of silica gel disposal.
Q: What yield improvements can be expected for the key intermediate?
A: Prior art methods for synthesizing the key mono-ester intermediate typically achieved yields of only 40-47%. The novel selective hydrolysis and amidation strategy described in patent CN102548958A boosts this yield to approximately 75%, significantly enhancing overall process efficiency and material throughput.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tricyclic Derivative Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of securing a stable supply of high-quality intermediates for oncology drug development. Our technical team has thoroughly analyzed the pathway described in CN102548958A and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production required to bring this chemistry to life. We are committed to delivering high-purity pharmaceutical intermediates that meet stringent purity specifications, supported by our rigorous QC labs equipped with state-of-the-art analytical instrumentation. Our facility is designed to handle complex organic syntheses safely, ensuring that the benefits of this safer, greener route are fully realized in the final product delivered to your doorstep.
We invite you to engage with our technical procurement team to discuss how we can support your specific project needs. Whether you require a Customized Cost-Saving Analysis for your current supply chain or need to verify specific COA data and route feasibility assessments for this tricyclic derivative, we are ready to provide the data-driven insights necessary for your decision-making. Partner with us to leverage this advanced synthesis technology and secure a competitive advantage in the global pharmaceutical market through superior quality and reliability.