Advanced Chemical Synthesis of Beta-Arbutin for High-Purity Cosmetic Intermediates
Advanced Chemical Synthesis of Beta-Arbutin for High-Purity Cosmetic Intermediates
The global demand for high-purity beta-arbutin, a premier skin-whitening agent and potential blood antifreeze, necessitates robust and scalable manufacturing protocols that transcend traditional limitations. Patent CN108997454B introduces a transformative chemical synthesis method that addresses critical bottlenecks in the production of this valuable fine chemical intermediate. By leveraging a novel fluoroglucose donor strategy, this technology eliminates the reliance on hazardous hydrogen bromide and unstable bromosugar intermediates that have historically plagued the industry. For R&D directors and supply chain leaders, this represents a pivotal shift towards safer, more efficient, and economically viable production pathways. The methodology ensures consistent stereochemical control, delivering the biologically active beta-anomer with exceptional purity, which is paramount for regulatory compliance in cosmetic and pharmaceutical applications.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the industrial synthesis of beta-arbutin has been constrained by significant safety and efficiency challenges inherent to older glycosylation strategies. Traditional routes often rely on the reaction of beta-pentaacetylglucose with hydrogen bromide in glacial acetic acid to generate alpha-bromoperacetylpyran-D-glucose, a highly unstable and toxic intermediate. This approach not only introduces severe occupational health hazards due to the handling of corrosive HBr gas but also suffers from poor atom economy and difficult waste management. Furthermore, when hydroquinone is employed directly as the glycosyl acceptor, the reaction frequently yields undesirable double-glycosidation by-products, drastically reducing the isolation yield of the target mono-glycoside. Even alternative methods using 4-hydroxyphenyl acetate often struggle with reduced nucleophilicity of the phenolic hydroxyl group, capping overall yields at suboptimal levels that fail to meet the rigorous cost targets of modern high-volume manufacturing.
The Novel Approach
In stark contrast, the innovative pathway detailed in the patent utilizes a tetra-acetyl-alpha-fluoroglucose donor, generated via mild fluorination with pyridine hydrofluoric acid, which offers superior stability and reactivity profiles compared to its brominated counterparts. This method strategically employs p-hydroxyacetophenone as the glycosyl acceptor, effectively blocking the para-position to prevent double glycosylation during the coupling step, thereby ensuring high regioselectivity. Following the glycosylation, a Baeyer-Villiger oxidation seamlessly converts the acetyl group into the requisite acetoxy functionality, which is subsequently deprotected to reveal the final phenolic structure. This multi-step sequence operates under remarkably mild thermal conditions, typically between 10°C and 30°C, minimizing energy consumption and thermal degradation risks. 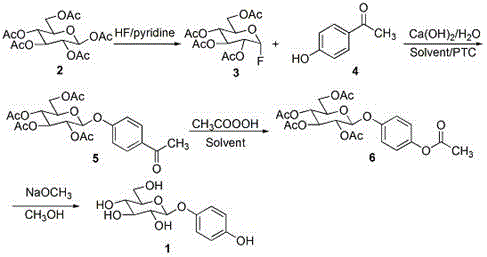
Mechanistic Insights into Fluoroglucose-Mediated Glycosylation
The cornerstone of this synthesis lies in the stereoselective glycosylation step where tetra-acetyl-alpha-fluoroglucose reacts with p-hydroxyacetophenone under phase transfer catalysis. The use of calcium hydroxide as a promoter alongside tetrabutylammonium bromide facilitates the generation of the phenoxide nucleophile in the organic phase, enhancing its reactivity towards the anomeric carbon of the fluorosugar. The fluorine atom at the anomeric position serves as an excellent leaving group, activated by the neighboring acetyl groups, which promotes the formation of the beta-glycosidic bond through a concerted SN2-like mechanism or an ion-pair pathway that favors the equatorial attack. This mechanistic precision is critical for avoiding the formation of the alpha-anomer, which lacks the desired biological activity, thus simplifying downstream purification and boosting the effective yield of the active pharmaceutical ingredient. The choice of p-hydroxyacetophenone is particularly ingenious; it acts as a temporary protecting group for the phenol, preventing over-reaction while maintaining sufficient nucleophilicity for efficient coupling.
Following the glycosylation, the transformation of the acetyl moiety to the acetoxy group via peracetic acid oxidation demonstrates a sophisticated application of the Baeyer-Villiger reaction within a carbohydrate context. This step proceeds with high fidelity, migrating the aryl group to the oxygen atom without disturbing the sensitive glycosidic linkage or the ester protecting groups on the sugar ring. The subsequent deprotection using sodium methoxide in anhydrous methanol is a classic Zemplén deacetylation, which cleanly removes all acetyl groups to furnish the final polyhydroxylated beta-arbutin structure. Impurity control is inherently built into this route; by avoiding the formation of di-glycosides and unstable halogenated intermediates, the crude product profile is significantly cleaner, reducing the burden on crystallization and chromatography units. 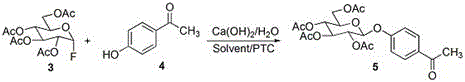
How to Synthesize Beta-Arbutin Efficiently
The operational simplicity of this four-step sequence makes it highly attractive for process chemists aiming to establish a reliable supply chain for beta-arbutin. The protocol begins with the fluorination of commercially available penta-acetyl-beta-D-glucose, followed by the key coupling reaction, oxidation, and final deprotection. Each step has been optimized to proceed at near-ambient temperatures, reducing the need for complex heating or cooling infrastructure. The detailed standardized synthesis steps, including specific molar ratios, solvent choices, and workup procedures, are outlined below to facilitate immediate technology transfer and pilot scale validation.
- React penta-acetyl-beta-D-glucose with 70% pyridine hydrofluoric acid at 10-30°C to obtain tetra-acetyl-alpha-fluoroglucose.
- Perform glycosylation of tetra-acetyl-alpha-fluoroglucose with p-hydroxyacetophenone using calcium hydroxide and tetrabutylammonium bromide at 20-30°C.
- Oxidize the acetyl group to an acetoxy group using 40% peracetic acid in an organic solvent at 5-20°C.
- Deprotect the acetyl groups using sodium methoxide in anhydrous methanol at 15-25°C to yield final beta-arbutin.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the adoption of this synthesis route offers profound strategic benefits that extend beyond mere technical feasibility. The elimination of toxic hydrogen bromide and unstable bromosugar intermediates significantly reduces the regulatory burden and safety costs associated with hazardous material handling and storage. This translates directly into a more resilient supply chain, as the reliance on specialized, high-risk reagents is minimized in favor of stable, commodity-grade chemicals like p-hydroxyacetophenone and peracetic acid. Furthermore, the high yields reported in each step—often exceeding 90%—imply a substantial reduction in raw material consumption per kilogram of final product, driving down the variable cost of goods sold without compromising on quality standards.
- Cost Reduction in Manufacturing: The process achieves significant cost optimization by maximizing atom economy and minimizing waste generation. By avoiding the formation of double-glycosidation by-products, the need for extensive purification to remove these structurally similar impurities is drastically reduced, lowering solvent usage and processing time. The use of calcium hydroxide as a low-cost promoter instead of expensive heavy metal catalysts further contributes to a leaner cost structure, making the final beta-arbutin product more competitive in the global marketplace while maintaining high purity specifications required for cosmetic grade applications.
- Enhanced Supply Chain Reliability: Sourcing stability is greatly improved as the key starting materials, such as penta-acetyl-glucose and p-hydroxyacetophenone, are widely available from multiple global suppliers, mitigating the risk of single-source bottlenecks. The mild reaction conditions (10-30°C) allow for production in standard stainless steel reactors without the need for exotic metallurgy or extreme temperature control systems, enabling flexible manufacturing across different geographic locations. This operational flexibility ensures consistent delivery schedules and protects downstream customers from volatility caused by equipment downtime or specialized reagent shortages.
- Scalability and Environmental Compliance: The synthesis is designed with green chemistry principles in mind, generating significantly less three-waste emission compared to traditional bromination methods. The absence of heavy metal residues simplifies wastewater treatment protocols and reduces the environmental footprint of the manufacturing site. This alignment with stringent environmental regulations facilitates smoother permitting processes for capacity expansion, allowing manufacturers to scale from pilot batches to multi-ton commercial production with confidence and minimal ecological impact.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this beta-arbutin synthesis technology. These insights are derived directly from the patent data to provide clarity on process robustness, yield expectations, and scalability factors that are critical for decision-makers evaluating this route for long-term procurement contracts.
Q: Why is p-hydroxyacetophenone used instead of hydroquinone in this synthesis?
A: Using hydroquinone directly often leads to double glycosylation by-products, reducing yield. P-hydroxyacetophenone acts as a protected acceptor that prevents this side reaction, ensuring higher selectivity for the mono-glycoside before oxidation converts it to the final phenol structure.
Q: What are the yield advantages of this fluoroglucose method?
A: This method achieves step yields exceeding 92-96% for key intermediates, significantly higher than traditional methods which often struggle to exceed 60% overall yield due to instability of bromosugar intermediates and side reactions.
Q: Is this process suitable for large-scale industrial production?
A: Yes, the process operates under mild conditions (10-30°C), avoids highly toxic hydrogen bromide gas, and generates less three-waste emission, making it environmentally compliant and safer for commercial scale-up.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Beta-Arbutin Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of securing a stable supply of high-quality cosmetic and pharmaceutical intermediates. Our technical team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory innovation to industrial reality is seamless. We are committed to delivering beta-arbutin that meets stringent purity specifications, supported by our rigorous QC labs that employ advanced analytical techniques to verify identity and assay. Our capability to implement this advanced fluoroglucose-based synthesis ensures that we can offer a product with a superior impurity profile, catering to the most demanding international regulatory standards.
We invite you to collaborate with us to optimize your supply chain for beta-arbutin and related fine chemicals. Contact our technical procurement team today to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are prepared to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing capabilities can enhance your product competitiveness and ensure uninterrupted supply continuity for your global operations.