Advanced Decitabine Manufacturing: Overcoming Isomer Separation Challenges for Commercial Scale
Advanced Decitabine Manufacturing: Overcoming Isomer Separation Challenges for Commercial Scale
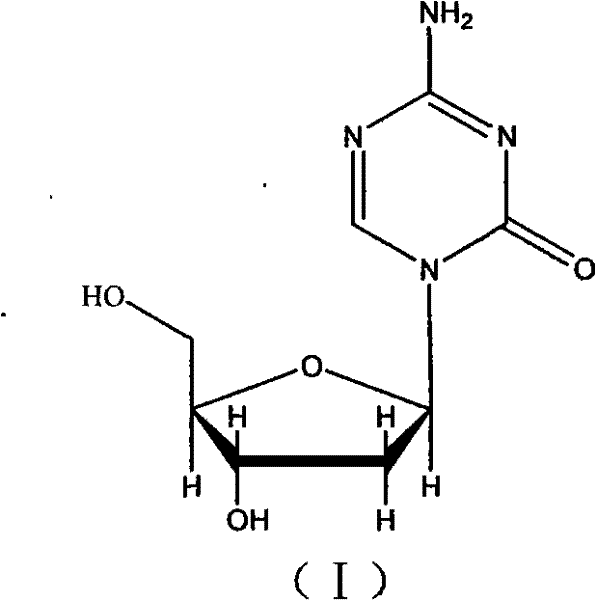
The pharmaceutical industry constantly seeks robust synthetic routes for oncology APIs that balance high purity with economic viability. Patent CN102010455B introduces a transformative method for preparing Decitabine (NSC 127716), a potent hypomethylating agent used in the treatment of myelodysplastic syndromes. Unlike conventional approaches that struggle with stereochemical control, this invention utilizes 4-amino-1-(β-D-erythro ribofuranose)-1,3,5-triazine-2(1H)-ketone as a strategic starting material. By employing a sequence of etherification, condensation, and selective deprotection, the process achieves a final purity exceeding 99.85 percent under mild reaction conditions. This technical breakthrough effectively bypasses the notoriously difficult separation of alpha and beta isomers, offering a streamlined pathway that is highly attractive for industrial scale-up and reliable supply chain integration.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the synthesis of Decitabine has been plagued by stereochemical inefficiencies that drive up costs and complicate manufacturing. Traditional routes often begin with 2-deoxy-D-ribose or protected ribose derivatives, which inevitably generate a mixture of alpha and beta anomers during the glycosylation or coupling steps. Separating these isomers is a significant bottleneck; the alpha-anomer is biologically inactive, yet its physical properties are often similar enough to the active beta-form to require repetitive chromatography or crystallization. Furthermore, prior art methods, such as those utilizing tin tetrachloride catalysis or acetylation strategies, frequently involve harsh conditions that can degrade the sensitive triazine ring. The cumulative effect of these limitations is a low overall yield, excessive solvent consumption, and a final product that requires rigorous purification to meet regulatory impurity profiles, thereby inflating the cost of goods sold for this critical cancer therapy drug.
The Novel Approach
The methodology disclosed in CN102010455B represents a paradigm shift by reversing the synthetic logic to preserve stereochemistry from the outset. Instead of constructing the nucleoside from a sugar and base separately, this approach starts with a pre-formed ribonucleoside derivative that already possesses the correct beta-configuration. The core innovation lies in the selective removal of the 2-hydroxyl group (deoxygenation) rather than the construction of the 2-deoxy bond. This strategy completely avoids the formation of the unwanted alpha-isomer, rendering the difficult separation step obsolete. The process utilizes mild reagents such as tetraisopropyldisiloxane derivatives for protection and radical mediators for deoxygenation, ensuring that the delicate glycosidic bond remains intact. This concise synthesis not only simplifies the operational workflow but also significantly enhances the theoretical yield, making it a superior candidate for cost reduction in pharmaceutical intermediates manufacturing.
Mechanistic Insights into Barton-McCombie Deoxygenation Strategy
The heart of this synthetic route is the application of the Barton-McCombie deoxygenation reaction, a powerful tool for converting alcohols to alkanes under radical conditions. In Step 2 and Step 3 of the process, the 2-hydroxyl group of the protected ribose intermediate is first activated by conversion into a thiocarbonate derivative using phenoxy thiocarbonyl chloride. This activation is crucial as it transforms a poor leaving group (hydroxyl) into a species capable of undergoing homolytic cleavage. Subsequently, in the presence of a radical initiator like AIBN and a hydrogen donor such as tributyltin hydride, a radical chain reaction is initiated. The tributyltin radical abstracts the sulfur-bound oxygen, generating a carbon-centered radical at the 2-position of the sugar ring. This radical then abstracts a hydrogen atom from the tin hydride, completing the deoxygenation. This mechanism is exceptionally valuable because it proceeds under neutral conditions, avoiding the acid-catalyzed hydrolysis or epimerization that often plagues nucleoside chemistry.
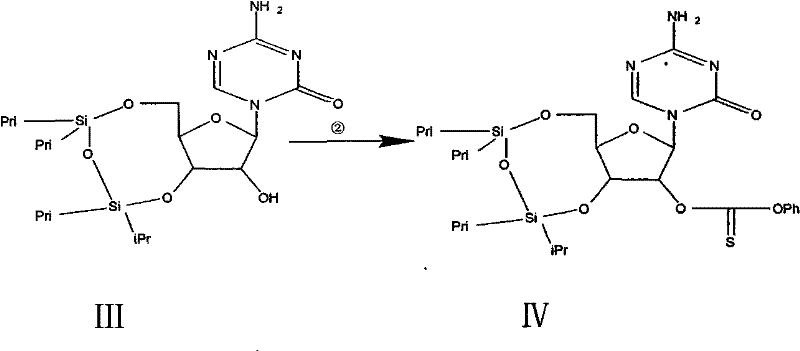
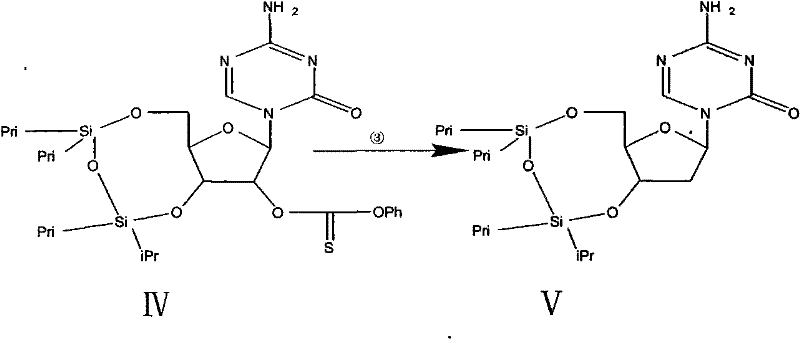
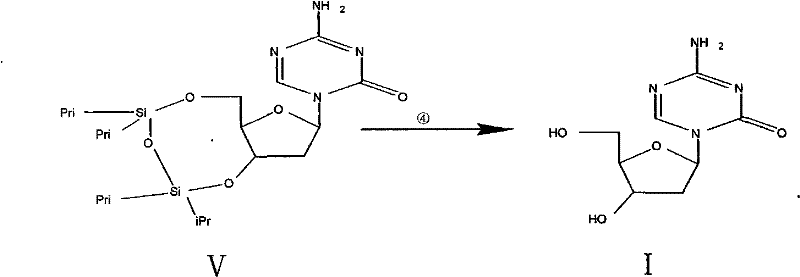
Impurity control in this process is inherently managed by the specificity of the radical mechanism and the choice of protecting groups. The use of the 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (TIPDS-Cl2) group in Step 1 provides robust protection for the 3 and 5 hydroxyls, preventing side reactions at these positions during the harsh radical step. Because the starting material is stereochemically pure (beta-configuration), and the radical intermediate at C2 is planar but constrained by the ring structure, the reformation of the C-H bond predominantly retains the desired stereochemistry or results in the thermodynamically stable product without generating the alpha-anomer impurity. The final deprotection step using tetrabutylammonium fluoride (TBAF) is highly selective for silicon-oxygen bonds, cleanly removing the protecting groups without affecting the triazine base. This orthogonal protection strategy ensures that the final crude product is already of high quality, requiring only a simple recrystallization to achieve the reported purity of greater than 99.85 percent.
How to Synthesize Decitabine Efficiently
The synthesis of Decitabine via this patented route involves four distinct chemical transformations that prioritize yield and stereochemical integrity. The process begins with the protection of the starting ribonucleoside, followed by activation of the 2-hydroxyl group, radical deoxygenation, and final global deprotection. Each step has been optimized for scalability, utilizing common organic solvents and reagents that are readily available in the global chemical market. The detailed standardized synthesis steps, including specific molar ratios, temperature controls, and workup procedures, are outlined in the guide below to assist process chemists in replicating this high-efficiency route.
- Protect the 3,5-hydroxyl groups of the ribose starting material using 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (TIPDS-Cl2) in anhydrous pyridine to form Intermediate III.
- Activate the 2-hydroxyl group by reacting Intermediate III with phenoxy thiocarbonyl chloride and DMAP in acetonitrile to generate the thiocarbonate Intermediate IV.
- Perform radical deoxygenation using tributyltin hydride and AIBN in refluxing hexane to remove the 2-oxygen functionality, yielding Intermediate V.
- Remove the silyl protecting groups using tetrabutylammonium fluoride (TBAF) in THF, followed by recrystallization to obtain pure Decitabine (>99.85% purity).
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers tangible strategic benefits beyond mere technical elegance. By eliminating the need for isomer separation, the process drastically reduces the number of unit operations required, which directly correlates to lower capital expenditure on equipment and reduced consumption of chromatography media and solvents. The avoidance of complex splitting processes means that production cycles are shorter, allowing for faster turnaround times and improved responsiveness to market demand fluctuations. Furthermore, the high purity achieved directly from the reactor minimizes the need for extensive reprocessing, leading to substantial cost savings in waste management and quality control testing. This efficiency makes the supply of high-purity pharmaceutical intermediates more reliable and economically sustainable.
- Cost Reduction in Manufacturing: The most significant economic driver of this technology is the elimination of the isomer separation step. In traditional synthesis, separating alpha and beta anomers often results in the loss of nearly half the theoretical yield, effectively doubling the cost of raw materials for the recovered portion. By starting with a stereochemically defined precursor and retaining that configuration throughout the synthesis, this method maximizes atom economy. Additionally, the use of mild reaction conditions reduces energy consumption associated with heating and cooling, while the simplified workup procedures decrease the volume of hazardous waste generated, further lowering disposal costs and enhancing the overall profit margin for commercial scale-up of complex oncology APIs.
- Enhanced Supply Chain Reliability: Supply chain resilience is bolstered by the robustness of the chemical steps involved. The reagents used, such as TIPDS-Cl2 and tributyltin hydride, are commodity chemicals with stable global availability, reducing the risk of raw material shortages. The process tolerance is high, meaning that minor variations in reaction parameters do not lead to catastrophic batch failures, ensuring consistent output quality. This reliability is critical for maintaining continuous production schedules for life-saving medications like Decitabine, where interruptions can have severe clinical consequences. The ability to produce material with >99.85 percent purity consistently ensures that downstream formulation partners receive material that meets strict regulatory specifications without delay.
- Scalability and Environmental Compliance: From an environmental and scaling perspective, this route is superior to older methods that rely on heavy metal catalysts or harsh acidic conditions. The radical deoxygenation step, while requiring tin reagents, is well-understood and manageable on a large scale with appropriate recycling protocols. The overall reduction in solvent usage and the avoidance of multiple crystallization steps reduce the facility's environmental footprint. This aligns with modern green chemistry principles and helps manufacturers meet increasingly stringent environmental regulations. The simplicity of the four-step sequence facilitates easy technology transfer from pilot plant to multi-ton commercial production, ensuring that the supply chain can scale rapidly to meet growing global demand for cancer therapies.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Decitabine synthesis method. These answers are derived directly from the experimental data and technical specifications provided in patent CN102010455B, offering clarity on process feasibility and product quality. Understanding these details is essential for stakeholders evaluating the potential for licensing or adopting this technology for their own manufacturing portfolios.
Q: How does this new method avoid the difficult separation of alpha and beta isomers?
A: Traditional methods start with 2-deoxy-D-ribose, which often yields a mixture of alpha and beta anomers requiring complex chromatographic separation. This patented method starts directly with 4-amino-1-(β-D-erythro ribofuranose)-1,3,5-triazine-2(1H)-ketone, which already possesses the correct beta-configuration. By selectively stripping the 2-hydroxyl group rather than building the sugar from scratch, the stereochemistry is preserved, eliminating the need for isomer splitting entirely.
Q: What is the achieved purity of Decitabine using this synthesis route?
A: The process described in patent CN102010455B achieves a final product purity of greater than 99.85% as detected by HPLC. This high level of purity is attained through the specificity of the radical deoxygenation step and a final recrystallization from methanol, ensuring the material meets stringent pharmaceutical standards without extensive purification cycles.
Q: Why is the Barton-McCombie deoxygenation preferred for this transformation?
A: The Barton-McCombie deoxygenation allows for the mild and selective removal of the secondary hydroxyl group at the 2-position under neutral radical conditions. Unlike acidic or basic hydrolysis methods which might degrade the sensitive triazine ring or cause epimerization, this radical mechanism proceeds with high chemoselectivity, preserving the integrity of the nucleobase and the glycosidic bond.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Decitabine Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and scalable synthesis routes for high-value oncology intermediates like Decitabine. Our team of expert process chemists has extensively analyzed the methodology described in CN102010455B and validated its potential for industrial application. We possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the transition from laboratory bench to manufacturing plant is seamless. Our state-of-the-art facilities are equipped with rigorous QC labs and advanced analytical instrumentation to guarantee stringent purity specifications, consistently delivering material that exceeds the 99.85 percent purity benchmark required for API synthesis.
We invite pharmaceutical companies and contract manufacturing organizations to collaborate with us to leverage this advanced technology. By partnering with our technical procurement team, you can access a Customized Cost-Saving Analysis tailored to your specific production volumes and regional regulatory requirements. We encourage you to contact us today to request specific COA data and route feasibility assessments, allowing us to demonstrate how our expertise in nucleoside chemistry can optimize your supply chain and reduce your overall cost of goods for Decitabine and related anticancer agents.