Industrial Scale-Up of High-Purity Baricitinib via Optimized Pivalate Protection Strategy
Introduction to Advanced Baricitinib Manufacturing
The global demand for Janus kinase (JAK) inhibitors continues to surge, driven by the critical need for effective treatments for rheumatoid arthritis and other autoimmune disorders. At the forefront of this therapeutic class is Baricitinib, a molecule whose complex heterocyclic structure has traditionally posed significant challenges for process chemists aiming for industrial viability. The recent disclosure in patent CN115181103A presents a transformative preparation method that addresses long-standing bottlenecks in yield, purity, and operational safety. This technical insight report analyzes the strategic shift from legacy SEM-protection strategies to a streamlined pivalate-based route, offering a compelling value proposition for procurement and supply chain leaders seeking a reliable baricitinib intermediate supplier.
The core innovation lies in the meticulous redesign of the protection and deprotection sequences. By utilizing methyl (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)pivalate as a starting material, the inventors have circumvented the hazardous and cumbersome steps associated with previous methodologies. The resulting process not only enhances the overall throughput but also ensures that the final active pharmaceutical ingredient meets the most stringent purity specifications, with the notorious hydroxymethyl intermediate impurity effectively suppressed to trace levels. This represents a significant leap forward in cost reduction in API manufacturing, aligning perfectly with the goals of modern generic and branded pharmaceutical production.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
To fully appreciate the technical merit of the new methodology, one must first examine the deficiencies inherent in the prior art, specifically the route disclosed in patent CN102026999B. The conventional synthesis relies heavily on the use of 2-(trimethylsilyl)ethoxymethyl chloride (SEMCl) to protect the pyrrolo[2,3-d]pyrimidine nitrogen. This approach is fraught with operational hazards and inefficiencies; it necessitates the use of sodium hydride (NaH) as a strong base, which introduces significant safety risks regarding hydrogen gas evolution and exothermic control on a large scale. Furthermore, the SEM group is notoriously difficult to remove, often requiring a two-step deprotection sequence that drags down the overall yield and complicates the purification workflow.
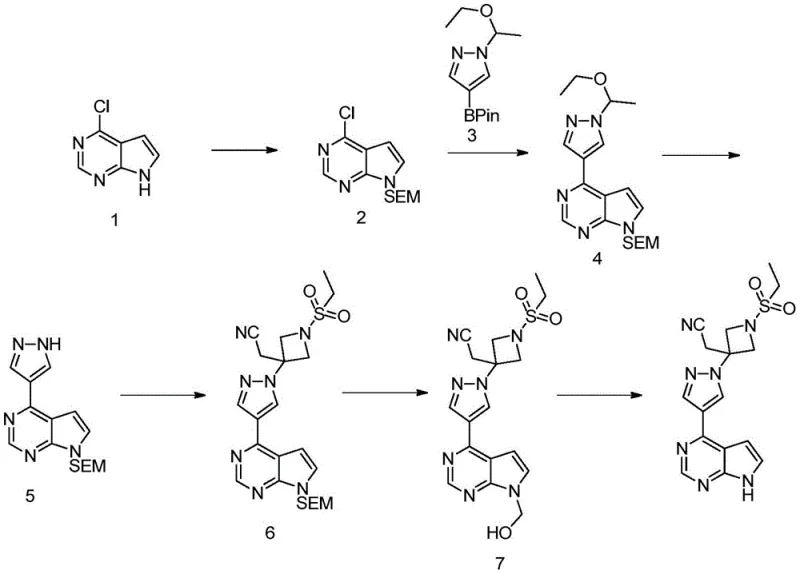
Beyond safety concerns, the economic implications of the legacy route are substantial. The SEM protecting group reagent is expensive, and the introduction of silicon atoms into the process stream creates downstream waste management challenges. Perhaps most critically, the literature route suffers from low overall yields and poor impurity profiles, particularly in the final hydrolysis step where the formation of hydroxymethyl byproducts is difficult to control. These factors combine to create a fragile supply chain that is vulnerable to batch failures and inconsistent quality, making it an unattractive option for high-volume commercial scale-up of complex heterocycles.
The Novel Approach
In stark contrast, the methodology outlined in CN115181103A introduces a robust and elegant solution by employing a pivalate protecting group strategy. The synthesis initiates with a Suzuki coupling reaction between the pivalate-protected chloropyrrolopyrimidine and a pyrazole boronate ester, establishing the core carbon-nitrogen bond with high efficiency. This is followed by a mild acidolysis step to reveal the pyrazole nitrogen, setting the stage for the subsequent Michael addition. The true brilliance of this route, however, is revealed in the final deprotection stage, where the pivalate group is cleaved under basic conditions that are far more forgiving and selective than those required for SEM removal.
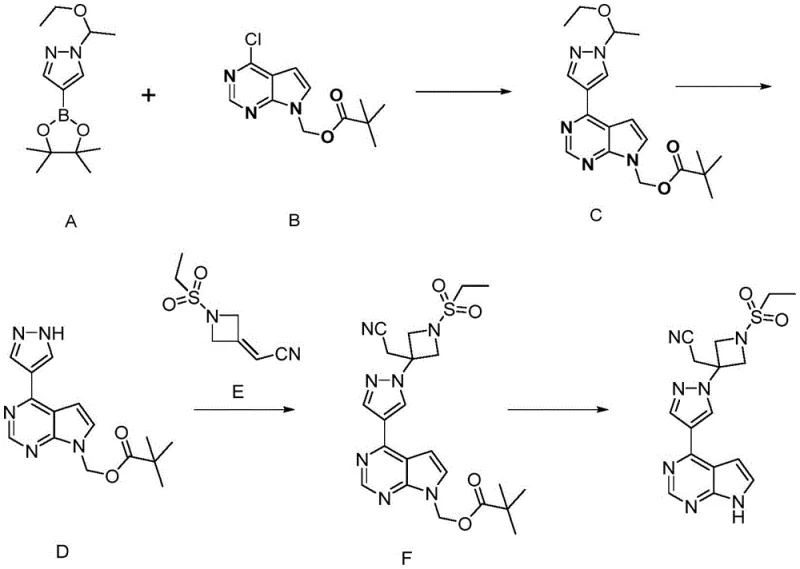
This novel approach fundamentally alters the economic and technical landscape of Baricitinib production. By eliminating the need for hazardous sodium hydride and expensive silyl reagents, the process inherently lowers the cost of goods sold (COGS). Moreover, the reaction conditions are optimized for scalability, utilizing common solvents and straightforward workup procedures such as filtration and crystallization. The transition from a silicon-based protection strategy to an oxygen-based pivalate strategy simplifies the molecular architecture of the intermediates, facilitating easier purification and ensuring a more consistent supply of high-quality intermediates for downstream formulation.
Mechanistic Insights into DBU-Catalyzed Hydrolysis and Impurity Control
A deep dive into the reaction mechanism reveals why this new route is superior for impurity management, specifically regarding the suppression of Impurity G. In traditional hydrolysis methods using inorganic bases like lithium hydroxide or sodium hydroxide, the nucleophilic attack on the ester linkage can be accompanied by side reactions that generate the hydroxymethyl intermediate (Compound 7 or Impurity G). This impurity is structurally similar to the target molecule, making it exceptionally difficult to separate via standard crystallization or chromatography, thereby threatening the final drug substance quality.
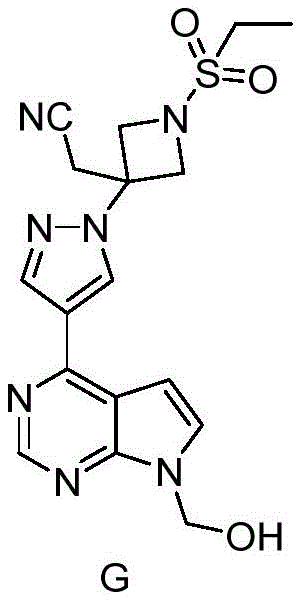
The patented process mitigates this risk through the strategic use of organic bases, such as 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU), for the hydrolysis step. The mechanistic advantage lies in the specific basicity and nucleophilicity profile of DBU, which facilitates the cleavage of the pivalate ester while minimizing the conditions that lead to the formation of the hydroxymethyl byproduct. Experimental data from the patent indicates that this optimization reduces Impurity G to below 0.15% in the crude product and further down to 0.02% after recrystallization. This level of control is critical for regulatory compliance and ensures that the final API possesses the requisite purity profile for clinical use without requiring resource-intensive purification steps.
Furthermore, the choice of solvent systems plays a pivotal role in the mechanistic success of this route. The use of THF and water mixtures for quenching and crystallization allows for precise control over the solubility of the intermediates. By adjusting the volume ratio and temperature, the process engineers can force the precipitation of the desired product while keeping impurities in solution. This thermodynamic control, combined with the kinetic selectivity of the DBU catalyst, creates a synergistic effect that maximizes yield and purity simultaneously, offering a textbook example of green chemistry principles applied to pharmaceutical manufacturing.
How to Synthesize Baricitinib Efficiently
The synthesis of Baricitinib via this novel pivalate route is designed for operational simplicity and high throughput. The process begins with the coupling of readily available starting materials, followed by a sequential deprotection and functionalization strategy that avoids the pitfalls of earlier methods. The key to success lies in the precise control of reaction parameters, particularly during the hydrolysis and crystallization phases, where the interplay between solvent composition and temperature dictates the final quality of the product. For process chemists looking to implement this technology, the following guide outlines the critical operational milestones derived from the patent examples.
- Perform Suzuki coupling between methyl (4-chloro-7H-pyrrolo[2,3-d]pyrimidin-7-yl)pivalate and a pyrazole boronate ester to form the coupled intermediate.
- Execute acidolysis to remove the ethoxyethyl protecting group, yielding the free pyrazole intermediate.
- Conduct a Michael addition with an azetidine acetonitrile derivative followed by DBU-catalyzed alkaline hydrolysis to remove the pivalate group and finalize the API.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this new synthetic route offers tangible benefits that extend far beyond mere chemical elegance. The primary advantage is the significant simplification of the supply chain for raw materials. By replacing specialized and costly reagents like SEMCl with commodity chemicals such as chloromethyl pivalate derivatives, the process reduces dependency on niche suppliers and mitigates the risk of raw material shortages. This shift not only stabilizes the supply chain but also creates opportunities for bulk purchasing discounts, directly impacting the bottom line through substantial cost savings in precursor acquisition.
- Cost Reduction in Manufacturing: The elimination of hazardous reagents like sodium hydride removes the need for specialized handling equipment and rigorous safety protocols associated with pyrophoric materials. This translates to lower operational expenditures (OPEX) regarding safety infrastructure and waste disposal. Additionally, the high yield and purity achieved in the final steps reduce the volume of solvent and energy required for purification, leading to a more efficient use of resources and a drastic simplification of the manufacturing workflow.
- Enhanced Supply Chain Reliability: The robustness of the pivalate protection strategy ensures consistent batch-to-batch quality, which is essential for maintaining uninterrupted production schedules. The process tolerance to minor variations in reaction conditions means that manufacturing campaigns are less likely to face delays due to out-of-specification results. This reliability is crucial for meeting the demanding delivery timelines of global pharmaceutical clients and securing long-term contracts for API supply.
- Scalability and Environmental Compliance: From an environmental perspective, the avoidance of silicon-based waste streams simplifies effluent treatment and aligns with increasingly strict environmental regulations. The process is inherently scalable, moving seamlessly from laboratory benchtop to multi-ton production without the need for complex engineering modifications. This scalability ensures that the supply of Baricitinib can be rapidly ramped up to meet market demand, providing a competitive edge in the fast-paced pharmaceutical landscape.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this novel Baricitinib synthesis route. These insights are derived directly from the experimental data and comparative examples provided in the patent documentation, offering clarity on how this technology compares to existing industry standards. Understanding these nuances is vital for technical teams evaluating the feasibility of technology transfer and for commercial teams assessing the value proposition of this manufacturing method.
Q: How does the new pivalate route improve impurity control compared to SEM protection?
A: The novel route replaces the troublesome SEM protecting group with a pivalate group. Crucially, the final deprotection uses organic bases like DBU instead of inorganic bases, which drastically reduces the formation of the difficult-to-remove hydroxymethyl intermediate (Impurity G) to below 0.15%.
Q: What are the key advantages for large-scale manufacturing of Baricitinib intermediates?
A: The process eliminates the need for hazardous sodium hydride (NaH) used in SEM protection and simplifies the deprotection sequence. The use of simple crystallization techniques (THF/Water) instead of column chromatography makes it highly suitable for multi-kilogram production.
Q: Can the final purity specifications meet stringent pharmaceutical standards?
A: Yes, the patent demonstrates that through recrystallization, the final Baricitinib product achieves a purity of 99.9%, with Impurity G content reduced to negligible levels (0.02%), meeting rigorous quality control requirements for active pharmaceutical ingredients.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Baricitinib Supplier
The technological advancements detailed in patent CN115181103A represent a paradigm shift in the production of high-value kinase inhibitors. NINGBO INNO PHARMCHEM stands ready to leverage this cutting-edge chemistry to deliver superior quality intermediates and APIs to the global market. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your project transitions smoothly from development to full-scale manufacturing. Our commitment to excellence is underpinned by stringent purity specifications and rigorous QC labs that guarantee every batch meets the highest international standards.
We invite you to collaborate with us to optimize your supply chain and reduce your time-to-market. Our technical team is prepared to provide a Customized Cost-Saving Analysis tailored to your specific production volumes and quality requirements. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments, and let us demonstrate how our advanced manufacturing capabilities can drive value for your organization.