Advanced Manufacturing of High-Purity Tolvaptan Intermediates for Global Pharmaceutical Supply Chains
The pharmaceutical landscape for cardiovascular therapeutics continues to evolve, with Tolvaptan standing out as a critical non-peptide selective antagonist for treating hyponatremia associated with congestive heart failure and liver cirrhosis. The commercial viability and clinical safety of this active pharmaceutical ingredient are intrinsically linked to the quality of its synthetic precursors, particularly the key intermediate known chemically as N-[4-[(7-chloro-2,3,4,5-tetrahydro-5-oxo-1H-1-benzazepin-1-yl)carbonyl]-3-methylphenyl]-2-methylbenzamide, often referred to as Formula II. Recent intellectual property developments, specifically patent CN108503586B, have unveiled a transformative approach to synthesizing this intermediate, addressing long-standing challenges regarding purity and impurity profiles that have plagued previous manufacturing attempts. This technical breakthrough is not merely an academic exercise but represents a significant leap forward for reliable pharmaceutical intermediates supplier networks seeking to enhance supply chain robustness. By achieving a purity level exceeding 99.00 percent and rigorously controlling specific genotoxic or process-related impurities like Impurity M to below 0.1 percent, this new methodology aligns perfectly with stringent International Council for Harmonisation (ICH) guidelines. For R&D Directors and Procurement Managers alike, understanding the nuances of this purification technology is essential for securing a stable, high-quality supply of Tolvaptan that meets global regulatory standards while optimizing production costs.
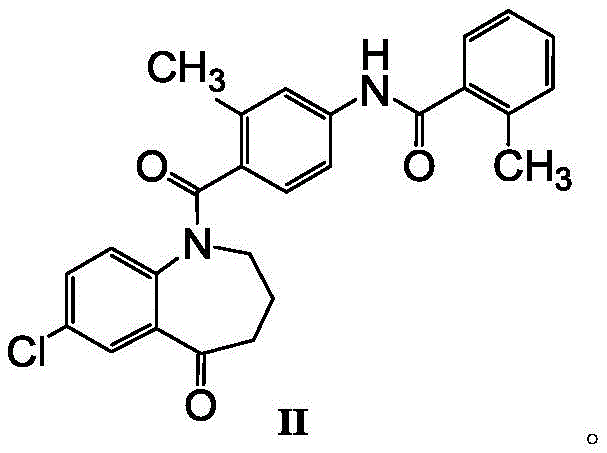
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the preparation of Tolvaptan intermediates has been fraught with inefficiencies that negatively impact both the economic and operational aspects of large-scale manufacturing. Prior art methodologies, such as those disclosed in earlier literature, often relied heavily on column chromatography for the purification of the crude intermediate. While column chromatography can theoretically achieve separation, it is notoriously unsuitable for industrial-scale production due to its excessive consumption of silica gel and solvents, high labor intensity, and significant difficulties in solvent recovery. Furthermore, alternative recrystallization techniques reported in patents like CN101817783A, which utilized methanol and isopropyl ether, frequently resulted in products with suboptimal purity levels ranging only between 97.5 percent and 98.8 percent. These conventional methods often failed to adequately remove stubborn process-related impurities, specifically Impurity M, which can persist in the final matrix and compromise the safety profile of the finished drug. The visual representation of these older synthetic routes highlights the complexity and the multiple steps involved that often lead to yield erosion.
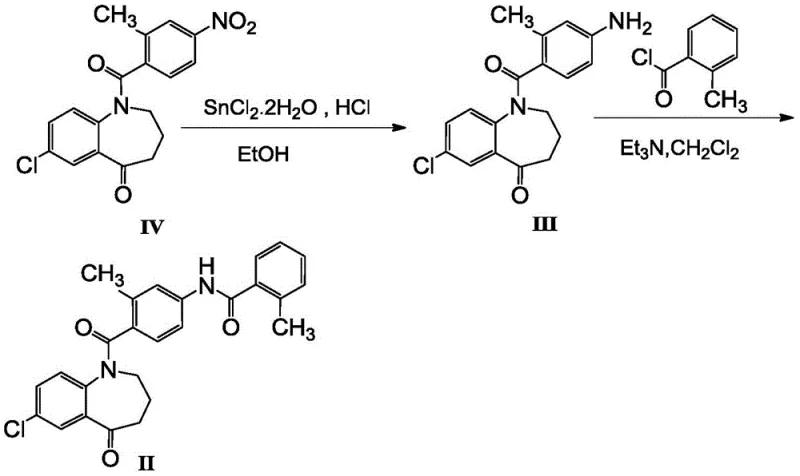
The Novel Approach
In stark contrast to these legacy processes, the innovative method described in patent CN108503586B introduces a highly efficient recrystallization protocol that bypasses the need for chromatographic purification entirely. This novel approach leverages a carefully engineered ternary solvent system comprising an ester, a halogenated alkane, and an ether. By optimizing the solubility parameters of the crude compound within this specific solvent mixture, the process facilitates the selective crystallization of the target intermediate while leaving impurities in the mother liquor. The result is a dramatic improvement in product quality, yielding a white solid with purity consistently above 99.00 percent, a significant upgrade from the yellowish or off-white solids produced by older methods. This shift from complex chromatographic separation to a streamlined crystallization process not only simplifies the operational workflow but also drastically reduces the environmental footprint and processing time. For supply chain stakeholders, this translates to a more predictable manufacturing timeline and a substantial reduction in the cost of goods sold, making the commercial scale-up of complex pharmaceutical intermediates far more economically attractive.
Mechanistic Insights into Amidation and Ternary Solvent Recrystallization
The core of this technological advancement lies in the precise control of the amidation reaction followed by the sophisticated thermodynamics of the recrystallization step. The synthesis begins with the reaction of 7-chloro-1,2,3,4-tetrahydro-1-(2-methyl-4-aminobenzoyl)-5H-1-benzazepin-5-one (Formula III) with o-methylbenzoyl chloride. This amidation is conducted in the presence of an acid-binding agent, such as pyridine or triethylamine, within a solvent system that may include dichloromethane or ethyl acetate. The reaction conditions are meticulously maintained between 20°C and 30°C to prevent side reactions that could generate difficult-to-remove byproducts. Following the formation of the crude Formula II, the critical purification occurs. The selection of ethyl acetate, dichloromethane, and isopropyl ether in a specific volume ratio, preferably around 1:4.5:1.5, creates a solvent environment where the solubility of the target molecule is highly temperature-dependent. Upon heating to 50°C to 60°C, the crude material dissolves completely, and upon controlled cooling, the target compound nucleates and grows into high-purity crystals. This mechanism effectively excludes Impurity M, which remains dissolved in the supernatant due to its different solubility profile in this specific ternary mixture.
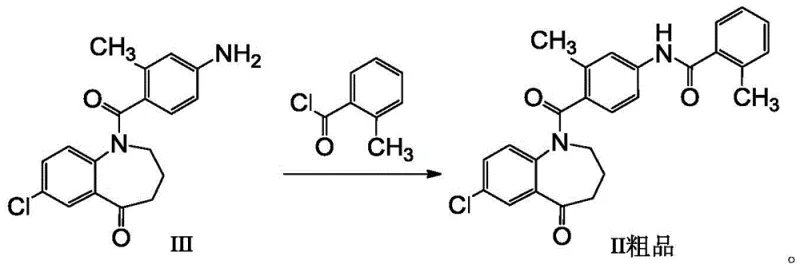
Furthermore, the control of impurity profiles is paramount for ensuring the safety of the final Tolvaptan API. Impurity M, a structural analog that can arise from incomplete reactions or side pathways, is particularly concerning due to potential toxicological implications. The new purification method demonstrates a remarkable capability to reduce the content of Impurity M to less than 0.1 percent, often achieving levels as low as 0.03 percent. This level of control is achieved not just by the solvent choice but also by the optimization of the solvent-to-solid ratio, typically maintained between 4:1 and 5:1. This ensures that there is sufficient solvent volume to dissolve impurities without sacrificing the yield of the desired product. The resulting intermediate is not only chemically pure but also physically superior, appearing as a white solid rather than the discolored products typical of less refined processes. This high degree of purity allows the intermediate to be carried forward into the final reduction step to produce Tolvaptan without requiring additional purification, thereby preserving the overall yield and integrity of the synthesis.
How to Synthesize Tolvaptan Intermediate Efficiently
To implement this advanced synthesis strategy effectively, manufacturers must adhere to a standardized protocol that emphasizes solvent quality and temperature control. The process begins with the preparation of the crude amidation product, ensuring that the reaction goes to completion before proceeding to purification. The subsequent recrystallization step is the critical control point where the majority of value is added. Operators must ensure the ternary solvent mixture is prepared with high precision, as deviations in the ratio of ester to halogenated alkane to ether can significantly impact the crystallization kinetics and final purity. Detailed standardized synthesis steps see the guide below.
- Perform amidation reaction between compound Formula III and o-methylbenzoyl chloride using an acid-binding agent in a suitable organic solvent.
- Dissolve the resulting crude compound Formula II in a heated mixture of ethyl acetate, dichloromethane, and isopropyl ether.
- Cool the solution to induce crystallization, followed by filtration and drying to obtain white solid with purity exceeding 99 percent.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented purification technology offers compelling strategic advantages that extend beyond simple technical metrics. The elimination of column chromatography represents a fundamental shift in the cost structure of manufacturing this intermediate. Chromatography is inherently expensive due to the high cost of stationary phases, the large volumes of solvents required, and the significant labor hours needed for operation and column packing. By replacing this with a scalable recrystallization process, manufacturers can achieve significant cost savings in pharmaceutical intermediates manufacturing. These savings are derived from reduced material consumption, lower waste disposal costs, and decreased energy usage associated with solvent recovery. Additionally, the simplified process flow reduces the risk of batch failures and cross-contamination, enhancing the overall reliability of the supply chain. This robustness is crucial for maintaining continuous supply to downstream API manufacturers who operate on tight schedules.
- Cost Reduction in Manufacturing: The transition from chromatographic purification to a ternary solvent recrystallization system eliminates the need for expensive silica gel and reduces solvent consumption drastically. This process optimization leads to substantial cost savings by minimizing waste generation and lowering the operational overhead associated with complex separation techniques. The higher yield obtained from this method, often exceeding 70 percent in purified form compared to lower yields in older methods, further contributes to a more favorable cost per kilogram of the final product. Consequently, this allows for more competitive pricing structures without compromising on the quality standards required for pharmaceutical applications.
- Enhanced Supply Chain Reliability: The simplified nature of the recrystallization process significantly reduces the lead time for high-purity pharmaceutical intermediates production. Unlike column chromatography, which can be a bottleneck due to its slow throughput and batch limitations, crystallization can be easily scaled up in standard reactor vessels. This scalability ensures that suppliers can respond more rapidly to fluctuations in market demand, providing a more stable and continuous supply of critical materials. The ability to consistently produce white, high-purity solids also reduces the likelihood of batch rejections during quality control testing, thereby smoothing out the logistics and inventory management for downstream partners.
- Scalability and Environmental Compliance: From an environmental and regulatory perspective, this method offers a greener alternative to traditional synthesis routes. The reduction in solvent usage and the avoidance of solid waste from chromatography columns align with modern green chemistry principles and increasingly strict environmental regulations. The process is inherently easier to scale from pilot plant quantities to multi-ton commercial production without losing efficiency or purity. This scalability ensures that the supply chain can grow alongside the market demand for Tolvaptan, supporting long-term commercial partnerships and reducing the risk of supply disruptions caused by capacity constraints or environmental compliance issues.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation and benefits of this specific purification technology. These insights are derived directly from the experimental data and technical specifications outlined in the patent documentation, providing clarity for stakeholders evaluating this process for integration into their supply chains. Understanding these details is crucial for making informed decisions about sourcing and manufacturing strategies.
Q: How does the new purification method improve impurity control compared to traditional column chromatography?
A: The novel recrystallization method utilizing a specific ternary solvent system effectively removes Impurity M to levels below 0.1 percent, whereas traditional column chromatography is labor-intensive and often yields lower purity with higher residual solvent risks.
Q: What are the specific solvent ratios required for the optimal recrystallization of Formula II?
A: Optimal results are achieved using a volume ratio of ester, halogenated alkane, and ether approximately at 1:4-5:1-2, specifically employing ethyl acetate, dichloromethane, and isopropyl ether to maximize yield and crystal quality.
Q: Can this intermediate be directly used for the final reduction step without further processing?
A: Yes, the high-purity intermediate obtained through this process possesses sufficient quality to undergo direct reduction to Tolvaptan, eliminating the need for additional purification steps and streamlining the overall manufacturing workflow.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tolvaptan Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the transition from laboratory innovation to commercial reality requires a partner with deep technical expertise and robust manufacturing capabilities. As a leading CDMO expert, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the promising results seen in patent literature are faithfully reproduced on an industrial scale. Our facilities are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch of Tolvaptan intermediate we supply meets the highest global standards. We understand the critical nature of Impurity M control and have optimized our processes to consistently deliver material that exceeds the 99 percent purity threshold, safeguarding your downstream API synthesis.
We invite you to collaborate with us to optimize your supply chain and reduce your overall manufacturing costs. Our technical procurement team is ready to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality needs. We encourage you to reach out to request specific COA data and route feasibility assessments to verify how our advanced purification technology can benefit your production line. By partnering with us, you gain access to a reliable source of high-quality intermediates that will support the consistent and compliant manufacturing of Tolvaptan for the global market.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →