Advanced Synthetic Route for Tolvaptan Intermediates Enabling Commercial Scale-Up
Advanced Synthetic Route for Tolvaptan Intermediates Enabling Commercial Scale-Up
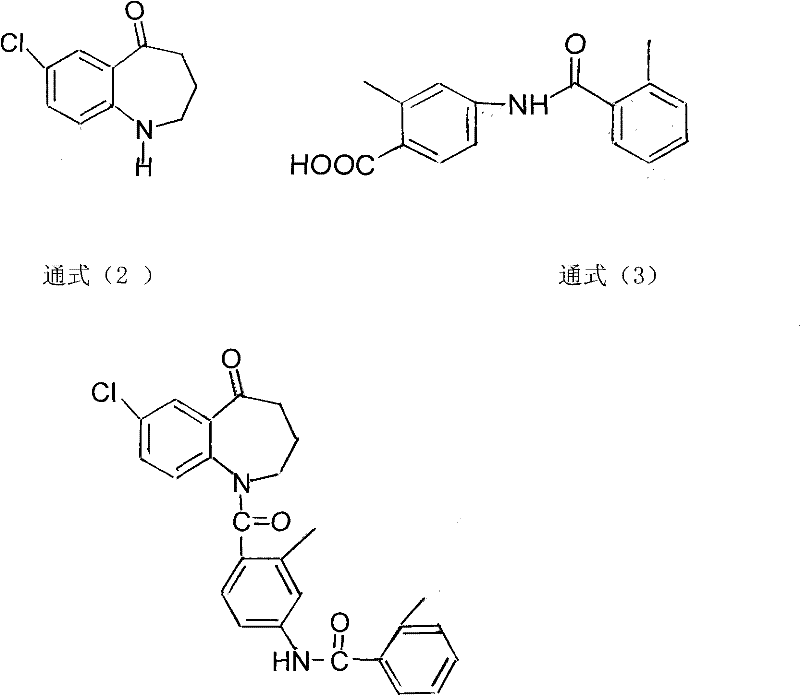
The pharmaceutical landscape for treating hyponatremia has been significantly advanced by the development of Tolvaptan, a non-peptide vasopressin V2-receptor antagonist. However, the commercial viability of such potent therapeutics relies heavily on the efficiency and purity of their synthetic precursors. Patent CN101817783B introduces a robust and innovative methodology for preparing the key Tolvaptan intermediate, 7-chloro-1-[2-methyl-4-(2-methylbenzoylamino)-benzoyl]-2,3,4,5-tetrahydro-1H-1-benzazepine-5-ketone. This technical disclosure addresses long-standing challenges in amidation reactions involving sterically hindered amines, offering a pathway that combines high yield with exceptional purity profiles. For R&D directors and process chemists, this patent represents a critical evolution in synthetic strategy, moving away from low-efficiency coupling agents toward a more direct and controllable acid chloride activation protocol.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior art methodologies for synthesizing this specific benzazepine derivative have historically struggled with significant inefficiencies that hinder large-scale production. As detailed in the background of the patent, traditional approaches such as the mixed anhydride method, active ester method, or carbodiimide coupling often result in unacceptably low transformation efficiencies, sometimes as low as 3% to 10%. The primary culprit is the substantial steric hindrance presented by the nitrogen atom within the seven-membered benzazepine ring of General Formula (2), which makes nucleophilic attack difficult. Furthermore, alternative strategies involving palladium-catalyzed carbonyl insertion, while conceptually elegant, necessitate complex post-reaction purification steps like column chromatography. In an industrial setting, the requirement for column chromatography is a severe bottleneck, complicating solvent recycling and drastically increasing operational costs, thereby rendering these methods unsuitable for the commercial scale-up of complex pharmaceutical intermediates.
The Novel Approach
The patented method overcomes these kinetic and thermodynamic barriers by employing a two-step sequence centered on the in situ formation of an acid chloride. By reacting General Formula (3) with thionyl chloride under precisely controlled thermal conditions, the process generates a highly reactive acylating agent that can effectively overcome the steric bulk of the amine partner. This approach eliminates the need for expensive coupling reagents like EDC or HATU and avoids the use of transition metal catalysts that require rigorous removal to meet regulatory standards. The result is a streamlined workflow that delivers the target intermediate with high purity directly from crystallization, bypassing the need for chromatographic purification entirely. This shift not only simplifies the unit operations but also significantly enhances the overall mass balance of the process.
Mechanistic Insights into Thionyl Chloride-Mediated Amidation
The core of this synthetic advancement lies in the optimization of the acid chloride formation step. The reaction between the carboxylic acid (General Formula 3) and thionyl chloride is not merely a standard activation; it is a kinetically controlled process where temperature plays a pivotal role in impurity management. The patent data reveals that maintaining the reaction temperature between 35°C and 45°C is critical. At lower temperatures, the reaction rate is insufficient, leading to prolonged reaction times that allow for the accumulation of side products. Conversely, excessive heat can degrade the sensitive benzazepine scaffold. By identifying this narrow thermal window, the inventors ensure that the acid chloride is generated rapidly and cleanly, ready for immediate consumption in the subsequent amidation step.
Following activation, the amidation reaction proceeds in the presence of an acid scavenger such as pyridine, n-butylamine, or DBU. The choice of base is crucial for neutralizing the hydrochloric acid byproduct without inducing racemization or degradation of the product. The mechanism involves the nucleophilic attack of the secondary amine nitrogen of General Formula (2) on the carbonyl carbon of the freshly formed acid chloride. Despite the steric congestion, the high electrophilicity of the acid chloride drives the reaction to completion. The use of aprotic solvents like dichloromethane or THF further facilitates this interaction by solvating the reactants effectively while remaining inert to the aggressive acylating conditions. This mechanistic understanding allows for precise control over the impurity profile, ensuring that the final product meets stringent HPLC purity specifications often exceeding 98%.
How to Synthesize 7-Chloro-1-benzazepine-5-ketone Efficiently
The implementation of this synthetic route requires careful attention to stoichiometry and thermal management to replicate the high yields reported in the patent embodiments. The process begins with the activation of the benzoic acid derivative using a slight excess of thionyl chloride, typically in a molar ratio of 1:1.2 to 1:5, depending on the specific solvent system employed. Following the removal of excess thionyl chloride, the crude acid chloride is coupled with the amine component at controlled low temperatures to manage exotherms. Detailed standardized synthetic steps see the guide below.
- Activate General Formula (3) by reacting with thionyl chloride in an aprotic solvent like THF or dioxane at 35-45°C for 3-6 hours to form the acid chloride solution.
- Perform amidation by adding the acid chloride solution to a mixture of General Formula (2) and an acid scavenger (e.g., pyridine or DBU) in dichloromethane at controlled temperatures below 25°C.
- Execute workup by quenching with ice water, adjusting pH to 2, extracting with organic solvents, and recrystallizing from methanol/isopropyl ether to achieve >97% HPLC purity.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the transition to this optimized synthetic route offers tangible benefits that extend beyond simple yield improvements. The elimination of chromatographic purification is perhaps the most significant economic driver, as it removes a batch-processing bottleneck that typically limits throughput and increases solvent consumption. By relying on crystallization for purification, the process becomes inherently more scalable and continuous-friendly, reducing the physical footprint required for manufacturing. This simplification of the downstream processing directly translates to substantial cost savings in both material usage and labor hours, making the supply of this critical intermediate more resilient to market fluctuations.
- Cost Reduction in Manufacturing: The replacement of expensive coupling reagents and palladium catalysts with commodity chemicals like thionyl chloride and pyridine drastically reduces the raw material cost per kilogram. Furthermore, the avoidance of column chromatography means that solvent recovery rates are significantly higher, as distillation is far more efficient than the disposal or complex recycling of chromatography fractions. This qualitative shift in the cost structure allows for a more competitive pricing model for the final API, providing a buffer against rising raw material costs in the global chemical market.
- Enhanced Supply Chain Reliability: The reliance on widely available solvents such as dichloromethane, THF, and toluene ensures that the supply chain is not vulnerable to shortages of specialized reagents. The robustness of the reaction conditions, which tolerate a reasonable range of temperatures and times without catastrophic failure, adds a layer of operational security. This reliability is crucial for maintaining consistent delivery schedules to downstream API manufacturers, reducing the risk of production delays that can ripple through the entire pharmaceutical supply network.
- Scalability and Environmental Compliance: From an environmental and safety perspective, the process is advantageous because it minimizes the generation of solid waste associated with silica gel from chromatography columns. The waste stream is primarily liquid, which is easier to treat and manage in standard effluent treatment plants. Additionally, the high selectivity of the reaction reduces the formation of hazardous byproducts, simplifying compliance with increasingly strict environmental regulations regarding chemical manufacturing emissions and waste disposal.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the scalability and purity of this specific Tolvaptan intermediate synthesis. These insights are derived directly from the experimental data and comparative analysis provided in the patent documentation, aimed at clarifying the operational advantages for potential manufacturing partners.
Q: How does this method improve upon traditional carbodiimide coupling for Tolvaptan intermediates?
A: Traditional carbodiimide methods often suffer from low transformation efficiency (around 10%) due to steric hindrance at the nitrogen atom of the benzazepine ring. This patented method utilizes an acid chloride intermediate formed under optimized thermal conditions (35-45°C), which significantly enhances reactivity and yield while minimizing difficult-to-remove impurities.
Q: What are the critical parameters for controlling impurities during the acid chloride formation?
A: The patent identifies temperature and reaction time as critical variables. Maintaining the acyl chloride reaction between 35-45°C for 3-6 hours prevents the generation of excessive impurities that occur at lower temperatures over extended periods or at excessively high temperatures. This precise control ensures the resulting acid chloride is sufficiently pure for the subsequent amidation step without requiring column chromatography.
Q: Is this synthetic route suitable for large-scale industrial production?
A: Yes, the method is explicitly designed for industrial scalability. By eliminating the need for column chromatography—a major bottleneck in previous methods—and utilizing common organic solvents like dichloromethane and THF, the process allows for straightforward workup and solvent recovery, making it highly viable for commercial scale-up of complex pharmaceutical intermediates.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Tolvaptan Intermediate Supplier
At NINGBO INNO PHARMCHEM, we recognize that the successful commercialization of complex therapeutics like Tolvaptan depends on a supply chain that prioritizes both quality and scalability. Our technical team has extensively analyzed the pathways described in CN101817783B and possesses the extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. We are equipped with rigorous QC labs and state-of-the-art pilot facilities capable of adhering to stringent purity specifications, ensuring that every batch of intermediate we produce meets the exacting standards required for GMP API synthesis.
We invite pharmaceutical partners to leverage our expertise to optimize their supply chains for this critical cardiovascular medication. By collaborating with us, you gain access to a Customized Cost-Saving Analysis tailored to your specific volume requirements. We encourage you to contact our technical procurement team today to request specific COA data and route feasibility assessments, ensuring a seamless transition from laboratory innovation to commercial reality.