Advanced Catalytic Activation Strategy for Commercial Scale-up of Complex Sofosbuvir Intermediates
The global demand for direct-acting antiviral agents, particularly for the treatment of Hepatitis C Virus (HCV), has necessitated the development of robust and highly efficient synthetic routes for key active pharmaceutical ingredients. Patent CN111116693B, published in April 2021, introduces a significant technological breakthrough in the preparation of Sofosbuvir, a nucleotide analog prodrug that has revolutionized HCV therapy. This patent discloses a novel method for preparing Sofosbuvir by catalyzing the activation of the sugar intermediate using a combination of 3,3-diaryl acrolein and a Grignard reagent. The structural complexity of Sofosbuvir, as illustrated in the chemical diagram below, requires precise stereochemical control and high purity to meet stringent regulatory standards for API manufacturing. 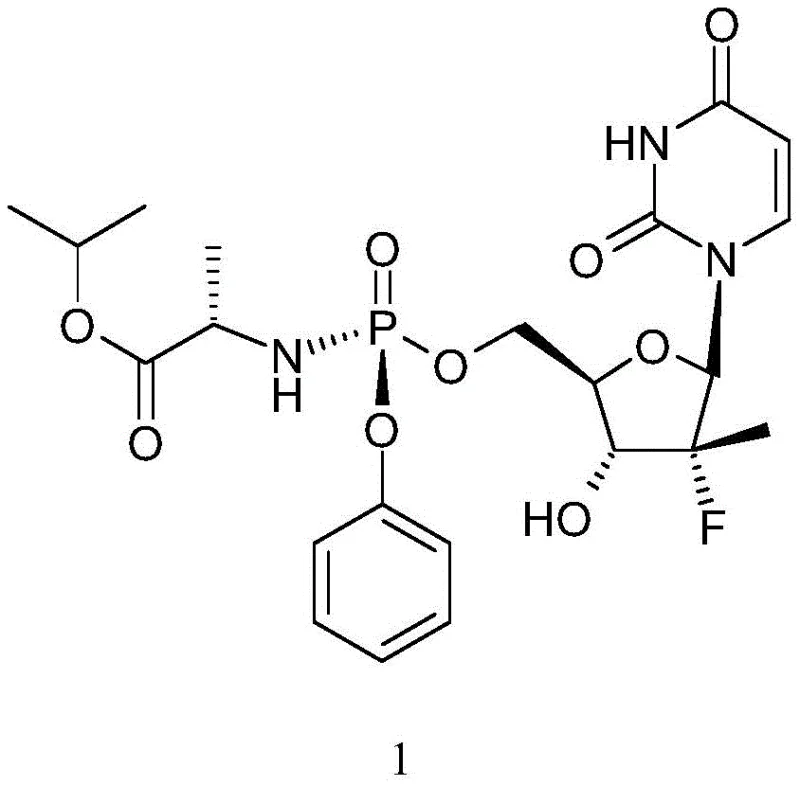 . By addressing the critical issue of disubstituted by-product formation, this innovation offers a pathway to significantly enhanced yield and purity profiles, making it a vital consideration for any reliable sofosbuvir intermediate supplier aiming to optimize their production capabilities.
. By addressing the critical issue of disubstituted by-product formation, this innovation offers a pathway to significantly enhanced yield and purity profiles, making it a vital consideration for any reliable sofosbuvir intermediate supplier aiming to optimize their production capabilities.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
In the conventional industrial synthesis of Sofosbuvir, the activation of the sugar precursor (Compound 2) is typically achieved using Grignard reagents such as tert-butyl magnesium chloride (t-BuMgCl). While this method is widely established, it suffers from a significant chemoselectivity defect. During the catalytic dehydrogenation process, the Grignard reagent does not exclusively target the hydroxyl group at the 5-position of the sugar ring; instead, it frequently removes the hydrogen from the 3-position hydroxyl group as well. This lack of selectivity leads to the formation of an intermediate species (Compound 5) which subsequently reacts with the phosphoramidate coupling partner (Compound 3) to generate a disubstituted by-product (Compound 6). 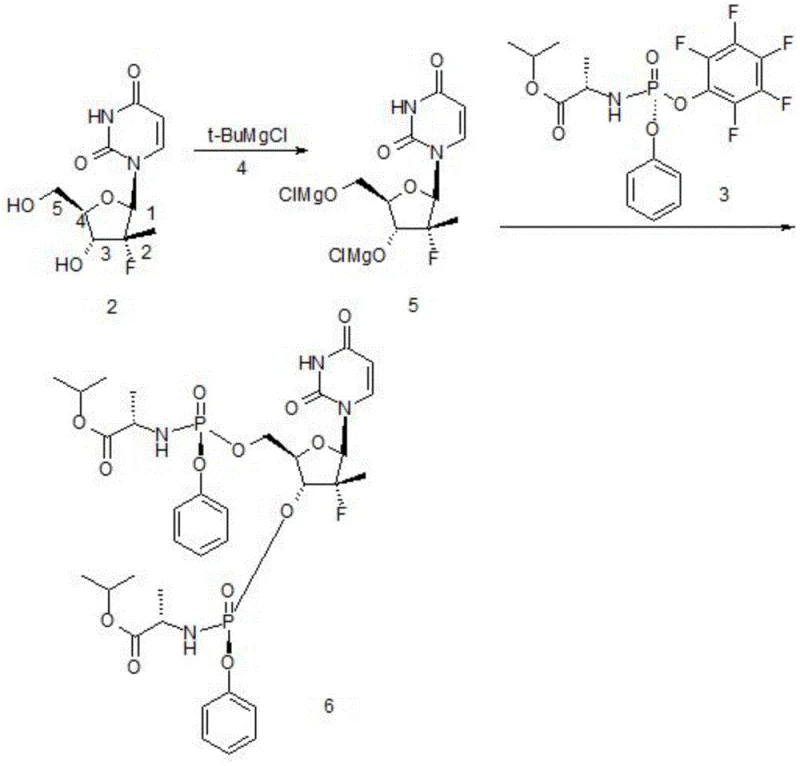 . The presence of this disubstituted impurity is detrimental to the overall process efficiency, as it not only consumes valuable starting materials but also complicates the downstream purification process. In comparative examples provided in the patent data, the conventional method resulted in a disubstituted by-product content of approximately 4.5%, which directly correlates to a lower isolated yield of 74.2% and necessitates more aggressive purification steps that can further erode material throughput.
. The presence of this disubstituted impurity is detrimental to the overall process efficiency, as it not only consumes valuable starting materials but also complicates the downstream purification process. In comparative examples provided in the patent data, the conventional method resulted in a disubstituted by-product content of approximately 4.5%, which directly correlates to a lower isolated yield of 74.2% and necessitates more aggressive purification steps that can further erode material throughput.
The Novel Approach
To overcome these inherent limitations, the patented method introduces a strategic modification to the activation step by incorporating 3,3-diarylacrolein as a co-catalyst or additive alongside the Grignard reagent. This novel approach fundamentally alters the reaction environment to favor the selective activation of the 5-position hydroxyl group while suppressing the unwanted reaction at the 3-position. The general reaction scheme demonstrates how Compound 2 is condensed with Compound 3 following this enhanced activation step to yield the target Sofosbuvir (Compound 1) with superior metrics. 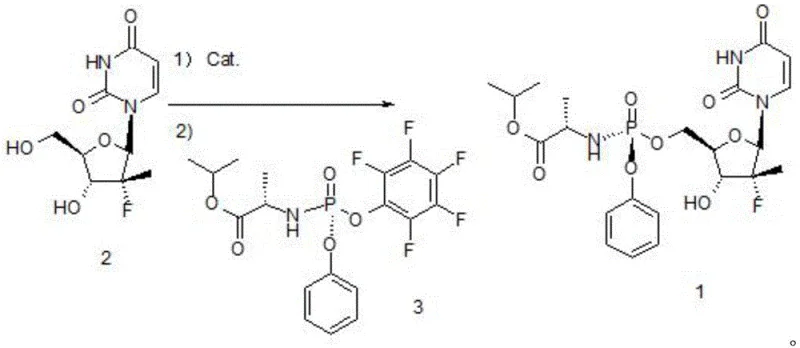 . Experimental data from the patent indicates that by utilizing 3,3-diphenylacrolein or its derivatives in a molar ratio of 0.05 to 0.2 relative to Compound 2, the formation of the disubstituted by-product is drastically reduced to as low as 0.8%. This improvement translates to a substantial increase in the isolated yield, reaching up to 86.2% with an HPLC purity exceeding 99.8%. This method represents a significant leap forward in cost reduction in API manufacturing, as it maximizes the conversion of expensive chiral intermediates into the final product while minimizing waste generation.
. Experimental data from the patent indicates that by utilizing 3,3-diphenylacrolein or its derivatives in a molar ratio of 0.05 to 0.2 relative to Compound 2, the formation of the disubstituted by-product is drastically reduced to as low as 0.8%. This improvement translates to a substantial increase in the isolated yield, reaching up to 86.2% with an HPLC purity exceeding 99.8%. This method represents a significant leap forward in cost reduction in API manufacturing, as it maximizes the conversion of expensive chiral intermediates into the final product while minimizing waste generation.
Mechanistic Insights into 3,3-Diarylacrolein Assisted Grignard Activation
The mechanistic advantage of this process lies in the interaction between the 3,3-diarylacrolein additive and the Grignard reagent within the tetrahydrofuran (THF) solvent system. It is hypothesized that the bulky diaryl groups of the acrolein derivative create a steric environment or a specific coordination complex with the magnesium species that shields the 3-position hydroxyl group of the sugar intermediate. When the t-BuMgCl is added dropwise at temperatures between -10°C and 0°C, the presence of the acrolein additive ensures that the deprotonation occurs selectively at the primary 5-hydroxyl group. This selectivity is critical because the 3-hydroxyl group is secondary and sterically more accessible in the absence of the additive, leading to the formation of the magnesium alkoxide at the wrong position. By preventing the formation of the 3-O-Mg intermediate, the subsequent nucleophilic attack on the phosphoramidate chloride (Compound 3) is restricted to the 5-position, thereby ensuring the correct regiochemistry of the final phosphoramidate bond. This level of control is essential for producing high-purity Sofosbuvir that meets the rigorous impurity profile requirements of global regulatory agencies.
Furthermore, the impurity control mechanism extends beyond just the activation step; it simplifies the entire workup and purification protocol. In the conventional process, the presence of significant amounts of the disubstituted by-product requires extensive chromatographic separation or multiple recrystallization cycles to achieve the necessary purity, which increases solvent consumption and processing time. In contrast, the novel method yields a crude product with significantly fewer structurally related impurities. The patent describes a streamlined purification process involving pH adjustment to 6-7 with dilute hydrochloric acid, solvent exchange, and a final reflux in dichloromethane followed by cooling crystallization. This process efficiently removes residual reagents and minor impurities, resulting in a white crystalline product with a melting point range of 95.8-98.6°C. The ability to achieve 99.7% to 99.8% purity directly from crystallization without complex chromatography is a major advantage for commercial scale-up of complex pharmaceutical intermediates, as it reduces the operational complexity and capital expenditure required for purification equipment.
How to Synthesize Sofosbuvir Efficiently
The synthesis of Sofosbuvir using this optimized protocol requires careful attention to reaction conditions and reagent stoichiometry to replicate the high yields reported in the patent. The process begins with the dissolution of the sugar intermediate (Compound 2) and the 3,3-diarylacrolein additive in anhydrous THF, followed by cooling to a precise temperature range of -5°C. The detailed standardized synthesis steps involve the controlled addition of the Grignard reagent and the subsequent coupling with the phosphoramidate, ensuring that the reaction kinetics favor the desired product. For R&D teams looking to implement this route, it is crucial to maintain the molar ratios specified, particularly the 1:1-2 ratio of Compound 2 to the Grignard reagent and the 1.1-1.5 ratio of Compound 3, to ensure complete conversion while minimizing excess reagent waste. The detailed standardized synthesis steps are outlined in the guide below.
- Dissolve Compound 2 and 3,3-diarylacrolein in THF, then cool the mixture to -10°C to 0°C under nitrogen protection.
- Dropwise add t-BuMgCl THF solution over 1 hour, maintaining temperature control to ensure selective activation of the 5-position hydroxyl group.
- Add Compound 3 solution dropwise, stir until HPLC shows less than 1% remaining Compound 2, then proceed to acidic workup and crystallization.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this patented synthesis route offers compelling economic and operational benefits that extend beyond simple yield improvements. The primary advantage lies in the significant cost reduction in API manufacturing driven by the enhanced atom economy of the process. By reducing the formation of the disubstituted by-product from 4.5% to less than 1%, the process ensures that a much higher proportion of the expensive chiral starting materials are converted into the saleable final product. This efficiency gain means that for every ton of raw material purchased, the output of finished Sofosbuvir is substantially higher, effectively lowering the cost of goods sold (COGS) without requiring a reduction in the purchase price of raw materials. Additionally, the simplified purification process reduces the consumption of solvents like ethyl acetate and dichloromethane, as well as the energy required for multiple distillation and crystallization cycles, contributing to a more sustainable and cost-effective manufacturing footprint.
- Cost Reduction in Manufacturing: The elimination of significant by-product formation directly translates to lower production costs per kilogram. Since the disubstituted impurity is structurally similar to the product, separating it is difficult and costly; by preventing its formation, the need for expensive preparative HPLC or multiple recrystallizations is removed. This qualitative improvement in the reaction profile allows manufacturers to operate with higher throughput and lower waste disposal costs, as fewer hazardous organic by-products are generated. The use of common reagents like t-BuMgCl and THF, combined with the catalytic amount of the acrolein additive, ensures that the raw material costs remain stable while the output value increases significantly.
- Enhanced Supply Chain Reliability: A more robust synthetic route with higher yields and fewer failure points enhances the overall reliability of the supply chain. Processes that generate high levels of impurities are prone to batch failures or the need for re-processing, which can lead to unpredictable lead times. By implementing this high-selectivity method, manufacturers can guarantee more consistent batch-to-batch quality and delivery schedules. This reliability is critical for downstream pharmaceutical companies that require a steady supply of high-purity Sofosbuvir intermediates to maintain their own production schedules for finished dosage forms. The reduced risk of batch rejection due to purity failures ensures a continuous flow of materials, reducing the need for safety stock and minimizing inventory holding costs.
- Scalability and Environmental Compliance: The process is designed with scalability in mind, utilizing standard unit operations such as dropwise addition, temperature-controlled stirring, and crystallization that are easily transferable from pilot plant to commercial scale. The reduction in by-product formation also has positive environmental implications, as it lowers the E-factor (mass of waste per mass of product) of the synthesis. This aligns with the increasing regulatory pressure on pharmaceutical manufacturers to adopt greener chemistry practices. The ability to scale this process from 100 kgs to 100 MT annual commercial production without significant modification makes it an ideal candidate for long-term supply agreements, ensuring that the supply chain can grow in tandem with market demand for Hepatitis C treatments.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis method. These answers are derived directly from the technical specifications and beneficial effects described in the patent literature, providing clarity on the operational parameters and expected outcomes. Understanding these details is essential for technical teams evaluating the feasibility of adopting this route for their own manufacturing processes or for procurement teams assessing the quality capabilities of potential suppliers.
Q: How does the addition of 3,3-diarylacrolein improve sofosbuvir purity?
A: The addition of 3,3-diarylacrolein during the Grignard activation step significantly suppresses the formation of the disubstituted by-product (Compound 6) by preventing the removal of hydrogen from the 3-position hydroxyl group, thereby increasing the HPLC purity to over 99.7%.
Q: What are the critical temperature controls for this synthesis?
A: The reaction requires precise thermal management, specifically cooling the initial mixture to -5°C during the dropwise addition of t-BuMgCl, followed by a controlled natural rise to 5°C for the activation period to ensure optimal selectivity.
Q: Is this method scalable for commercial API production?
A: Yes, the process utilizes standard solvents like THF and common reagents like t-BuMgCl, and the simplified purification steps involving dichloromethane reflux make it highly suitable for commercial scale-up from 100 kgs to 100 MT annual production.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Sofosbuvir Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of efficient and high-yielding synthetic routes in the competitive landscape of antiviral drug manufacturing. Our technical team has extensively analyzed the advancements presented in patent CN111116693B and possesses the expertise to implement this 3,3-diarylacrolein assisted activation strategy on a commercial scale. We have extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this patent are realized in actual manufacturing output. Our facilities are equipped with stringent purity specifications and rigorous QC labs capable of detecting impurities at trace levels, guaranteeing that every batch of Sofosbuvir intermediate we produce meets the highest global standards for pharmaceutical quality and safety.
We invite pharmaceutical companies and contract manufacturing organizations to collaborate with us to optimize their Sofosbuvir supply chain. By leveraging our technical capabilities and this advanced synthesis method, we can help you achieve significant operational efficiencies and cost savings. Please contact our technical procurement team to request a Customized Cost-Saving Analysis tailored to your specific volume requirements. We are ready to provide specific COA data and route feasibility assessments to demonstrate how our manufacturing excellence can support your product development and commercialization goals.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →