Scalable Synthesis of Fluoropyridazinone: A Technical Breakthrough for Commercial Manufacturing
The chemical landscape of heterocyclic fluorination has long been dominated by complex and often hazardous methodologies, yet the introduction of patent CN1583727A marks a significant paradigm shift in the production of fluoropyridazinone derivatives. This pivotal intellectual property outlines a robust nucleophilic substitution strategy that replaces traditional, energy-intensive routes with a streamlined solution-phase reaction. By leveraging the reactivity of halogenated pyridazinone precursors against readily available metal fluorides, this technology addresses the critical industry demand for high-purity intermediates essential for modern agrochemical and pharmaceutical applications. The method specifically targets the structural motifs found in biologically active compounds similar to fluorouracil, offering a versatile platform for generating diverse fluorinated scaffolds. For R&D directors and process chemists, this patent represents a validated pathway that balances reaction efficiency with operational safety, ensuring that the resulting fluoropyridazinone products meet stringent quality specifications required for downstream drug synthesis.
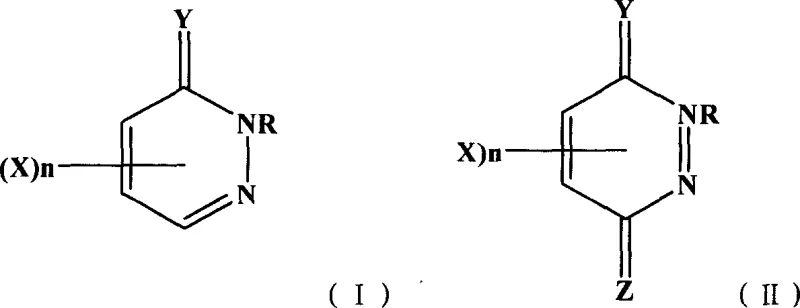
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Prior to the development of this innovative solution-phase technique, the industry relied heavily on three primary methods for introducing fluorine atoms into the pyridazine ring, each fraught with significant technical and economic drawbacks. The melting method, while historically common for polyfluorination, suffers from extremely poor selectivity, often yielding complex mixtures of perfluorinated byproducts that are notoriously difficult to separate. Furthermore, the requirement for high-temperature molten states poses severe safety risks and limits the functional group tolerance, making it unsuitable for synthesizing complex, sensitive intermediates. Alternatively, the diazotization method, though selective, is restricted to amino-substituted precursors and involves hazardous diazonium salts, limiting its scalability and industrial viability. Finally, condensation methods are inherently constrained by the availability and cost of specific hydrocarbon fluoride starting materials, creating a bottleneck in the supply chain for specialized fluoropyridazinones.
The Novel Approach
In stark contrast to these legacy techniques, the novel approach detailed in the patent utilizes a direct nucleophilic displacement of halogen atoms (chlorine, bromine, or iodine) with metal fluorides in a controlled liquid phase. This methodology allows for precise tuning of reaction parameters, such as temperature and solvent polarity, to achieve high yields without the formation of intractable side products. By employing aprotic polar solvents like dimethyl sulfoxide or acetone, the reaction medium effectively solvates the fluoride ion, enhancing its nucleophilicity while maintaining mild operating conditions typically between 30°C and 100°C. This shift from harsh thermal processes to温和 solution chemistry not only improves the safety profile but also enables the synthesis of a broader range of substituted derivatives, including those with sensitive alkyl or phenyl groups. The versatility of this approach is further evidenced by its ability to handle various leaving groups, providing manufacturers with flexibility in sourcing raw materials.
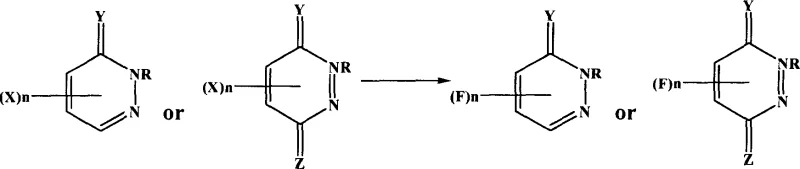
Mechanistic Insights into Metal Fluoride-Mediated Nucleophilic Substitution
The core mechanistic driving force behind this synthesis is a classic nucleophilic aromatic substitution (SnAr) facilitated by the electron-withdrawing nature of the pyridazinone ring system. In this catalytic cycle, the electron-deficient carbon atom bearing the halogen leaving group becomes susceptible to attack by the fluoride anion, which is activated by the polar aprotic solvent environment. The presence of carbonyl groups at the 6-position (or thio-carbonyl equivalents) significantly stabilizes the Meisenheimer complex intermediate, lowering the activation energy required for the substitution to proceed. This electronic activation allows even less reactive chloride leaving groups to be displaced efficiently under moderate heating, eliminating the need for exotic catalysts or extreme pressures. Understanding this electronic interplay is crucial for process optimization, as it dictates the stoichiometric ratios and temperature profiles necessary to drive the reaction to completion while minimizing hydrolysis or other degradation pathways.
From an impurity control perspective, the mechanism offers inherent advantages by avoiding the radical pathways often associated with high-temperature fluorination. The ionic nature of the substitution ensures that side reactions are largely limited to unreacted starting materials or simple hydrolysis products, which are easily removed during the aqueous workup described in the patent. The use of specific alkali metal fluorides, such as potassium fluoride, provides a consistent source of nucleophilic fluoride without introducing heavy metal contaminants that would require costly scavenging steps later in the process. Furthermore, the ability to recover and recycle the solvent post-reaction means that any soluble organic impurities can be concentrated and managed effectively, ensuring that the final crystallized product achieves the high purity levels demanded by regulatory bodies for pharmaceutical intermediates. This clean reaction profile translates directly into reduced downstream processing costs and higher overall process mass intensity.
How to Synthesize Fluoropyridazinone Efficiently
The synthesis protocol outlined in the patent provides a clear, reproducible framework for manufacturing fluoropyridazinone derivatives at scale, emphasizing simplicity and resource efficiency. The process begins with the careful selection of a halogenated precursor and an appropriate alkali metal fluoride, which are combined in an aprotic polar solvent at a optimized molar ratio to ensure complete conversion. Detailed standardized synthetic steps regarding specific charging orders, agitation speeds, and temperature ramping profiles are critical for maintaining batch-to-batch consistency and maximizing yield. Operators must adhere strictly to the recommended reaction times and temperatures to prevent over-fluorination or decomposition, leveraging the kinetic data provided in the experimental examples to fine-tune the process for specific substrates.
- Charge a reactor with halogenated pyridazinone and alkali metal fluoride (e.g., KF) in an aprotic polar solvent like DMSO or acetone.
- Heat the mixture to 30-100°C and stir for 3-10 hours to facilitate the nucleophilic substitution reaction.
- Recover the solvent via distillation, dissolve the residue in warm water, filter the solid, and recrystallize from ethanol to obtain pure product.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain leaders, the adoption of this synthesis route offers profound strategic benefits that extend far beyond simple chemical transformation. The primary value driver lies in the drastic simplification of the raw material portfolio; by utilizing commodity halogenated pyridazinones and inexpensive alkali metal fluorides, manufacturers can decouple their supply chains from volatile markets for specialized fluorinating reagents. This shift to abundant, low-cost starting materials significantly mitigates the risk of supply disruptions and price spikes, ensuring a stable and predictable cost base for long-term production planning. Additionally, the elimination of complex multi-step sequences or hazardous reagents reduces the operational overhead associated with safety compliance and waste disposal, further enhancing the economic viability of the process.
- Cost Reduction in Manufacturing: The economic model of this process is fundamentally superior due to the elimination of expensive transition metal catalysts and the ability to recycle the bulk solvent. By recovering solvents like DMSO or acetone through distillation, the facility drastically reduces its consumption of fresh chemicals, leading to substantial operational expenditure savings over time. Furthermore, the high selectivity of the reaction minimizes the loss of valuable starting materials to byproduct formation, thereby improving the overall atom economy and reducing the cost per kilogram of the final active intermediate. The avoidance of cryogenic conditions or high-pressure equipment also lowers capital expenditure requirements, making this technology accessible for both pilot and commercial-scale facilities.
- Enhanced Supply Chain Reliability: The reliance on widely available alkali metal fluorides and standard halogenated precursors creates a resilient supply chain that is less susceptible to geopolitical or logistical bottlenecks. Unlike specialized fluorinating agents that may have single-source suppliers, the reagents required for this synthesis are produced globally in massive quantities, ensuring continuous availability even during market fluctuations. This robustness allows procurement teams to negotiate better terms with multiple vendors and maintain healthy inventory levels without the fear of obsolescence. Moreover, the simplified logistics of handling solid fluorides and liquid solvents reduce the complexity of transportation and storage, streamlining the entire inbound supply chain operation.
- Scalability and Environmental Compliance: From an environmental and scaling perspective, this method aligns perfectly with modern green chemistry principles by minimizing waste generation and energy consumption. The ability to run reactions at moderate temperatures (30-100°C) significantly reduces the energy load compared to melting or high-temperature condensation methods, contributing to a lower carbon footprint for the manufacturing site. The closed-loop solvent recovery system ensures that volatile organic compound (VOC) emissions are kept to a minimum, facilitating easier compliance with increasingly stringent environmental regulations. This sustainability profile not only future-proofs the production asset but also enhances the brand reputation of the supplier as a responsible partner in the global pharmaceutical value chain.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this fluoropyridazinone synthesis technology. These answers are derived directly from the experimental data and claims within the patent documentation, providing a factual basis for decision-making. Understanding these nuances is essential for evaluating the feasibility of integrating this route into existing manufacturing workflows or for assessing the quality of potential suppliers.
Q: What are the advantages of this fluorination method over traditional melting methods?
A: Unlike traditional melting methods which often yield perfluorinated mixtures with poor selectivity and require extreme temperatures, this patented process utilizes mild conditions (30-100°C) in solution. This ensures high regioselectivity for mono- or di-fluorination, significantly simplifying downstream purification and improving overall product quality.
Q: Which metal fluorides are most effective for this synthesis?
A: The patent data indicates that alkali metal fluorides, particularly potassium fluoride (KF), sodium fluoride (NaF), and cesium fluoride (CsF), are highly effective. Potassium fluoride is noted as a preferred embodiment due to its balance of reactivity and cost-effectiveness in aprotic polar solvents.
Q: Can the solvents used in this process be recycled?
A: Yes, a key feature of this technology is the use of aprotic polar solvents such as dimethyl sulfoxide (DMSO), acetone, or acetonitrile, which can be recovered via reduced pressure distillation after the reaction. This recyclability drastically reduces waste generation and lowers the overall environmental footprint of the manufacturing process.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Fluoropyridazinone Supplier
As the global demand for fluorinated heterocycles continues to surge, partnering with a technically proficient manufacturer is critical for securing a competitive edge in the pharmaceutical and agrochemical sectors. NINGBO INNO PHARMCHEM stands at the forefront of this industry, leveraging deep expertise in nucleophilic substitution chemistry to deliver high-purity fluoropyridazinone intermediates that meet the most rigorous international standards. Our state-of-the-art facilities are equipped to handle complex synthetic pathways, boasting extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. We maintain stringent purity specifications through our rigorous QC labs, ensuring that every batch delivered is free from critical impurities and ready for immediate use in your downstream API synthesis.
We invite you to collaborate with us to optimize your supply chain and reduce your overall manufacturing costs through our advanced process technologies. Our technical team is prepared to provide a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality needs. We encourage you to contact our technical procurement team today to request specific COA data and comprehensive route feasibility assessments, allowing you to make informed decisions that drive value and efficiency for your organization.