Scalable Synthesis of Hexahydrofurofuranol Intermediates for Commercial API Production
Scalable Synthesis of Hexahydrofurofuranol Intermediates for Commercial API Production
The pharmaceutical industry continuously seeks robust synthetic pathways for critical antiretroviral agents, particularly those targeting HIV protease inhibition. A pivotal intermediate in this domain is hexahydrofuro[2,3-b]furan-3-ol, a core structural motif essential for the manufacture of Darunavir. Recent advancements documented in patent CN103864813A introduce a transformative methodology that addresses longstanding inefficiencies in producing this complex bicyclic system. This technical insight report analyzes the novel synthetic strategy, highlighting its potential to redefine supply chain stability and cost structures for global reliable pharmaceutical intermediate supplier networks. By shifting away from hazardous reagents and cryogenic dependencies, this approach offers a pragmatic solution for cost reduction in API manufacturing while maintaining the rigorous quality standards required for high-purity Darunavir intermediate production.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of the hexahydrofuro[2,3-b]furan scaffold has been plagued by significant operational hurdles that impede efficient commercial scale-up of complex pharmaceutical intermediates. Prior art methodologies frequently rely on the formation of nitromethyl intermediates, necessitating the use of nitromethane, a hazardous substance that imposes severe safety protocols and waste disposal burdens on manufacturing facilities. Furthermore, established routes often demand extreme cryogenic conditions, such as reactions maintained at -78°C, which are energy-intensive and technically challenging to sustain in large-volume reactors. The reliance on expensive chiral metal catalysts containing elements like Ytterbium or Scandium further escalates production costs, rendering many existing processes economically unviable for generic drug manufacturers seeking to optimize their supply chains without compromising on quality or regulatory compliance.
The Novel Approach
In stark contrast to these legacy challenges, the methodology outlined in CN103864813A presents a streamlined pathway characterized by operational simplicity and economic feasibility. This innovative route eliminates the need for dangerous nitro-compounds and avoids the logistical nightmare of maintaining ultra-low temperatures throughout the synthesis. Instead, it leverages readily available starting materials such as alkoxyacetates and 1,4-butyrolactone, which are accessible through standard chemical supply channels. By utilizing common bases like sodium tert-butoxide and standard reducing agents, the process significantly lowers the barrier to entry for production. This shift not only enhances safety profiles but also facilitates reducing lead time for high-purity API intermediates by simplifying the overall workflow, allowing manufacturers to respond more agilely to market demands for antiretroviral therapies.
Mechanistic Insights into Base-Catalyzed Substitution and Cyclization
The core of this synthetic breakthrough lies in a meticulously designed three-step sequence that constructs the bicyclic framework with high precision. The initial phase involves a base-catalyzed substitution reaction where an alkoxyacetate derivative reacts with 1,4-butyrolactone. This step is critical as it establishes the carbon backbone necessary for subsequent ring closure. Following this, a selective reduction transforms the ketone functionality into a hydroxyl group, setting the stage for the final cyclization. The concluding step employs a strong protic acid to induce intramolecular etherification, effectively locking the structure into the desired hexahydrofuro[2,3-b]furan-3-ol configuration. This mechanistic elegance ensures that side reactions are minimized, thereby preserving the integrity of the molecule throughout the synthesis.
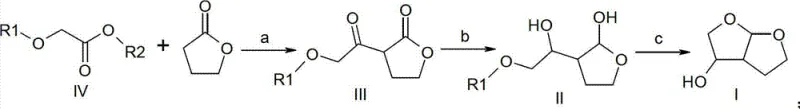
Controlling the impurity profile is paramount when synthesizing intermediates for potent pharmaceuticals like Darunavir. The patented process incorporates specific purification strategies, such as pH adjustments and solvent extractions, to remove residual starting materials and by-products effectively. For instance, the use of methyl tert-butyl ether as a solvent not only facilitates the reaction but also aids in the separation of organic phases during workup. Additionally, the selection of reducing agents like sodium borohydride allows for controlled reactivity, preventing over-reduction or degradation of sensitive functional groups. This attention to detail in the reaction design ensures that the final product meets the stringent purity specifications required by regulatory bodies, thereby reducing the risk of batch rejection and ensuring a consistent supply of high-purity Darunavir intermediate for downstream drug formulation.
How to Synthesize Hexahydrofuro[2,3-b]furan-3-ol Efficiently
Implementing this synthesis requires adherence to specific operational parameters to maximize yield and purity. The process begins with the preparation of the keto-lactone intermediate under inert atmosphere conditions to prevent moisture interference. Subsequent reduction steps must be carefully monitored to ensure complete conversion without generating excessive diastereomers. Finally, the acid-catalyzed cyclization requires precise temperature control to drive the equilibrium towards the desired bicyclic product. While the general chemistry is robust, scaling these steps requires specialized equipment and expertise to maintain consistency across large batches. The detailed standardized synthesis steps see the guide below for specific operational protocols.
- Perform base-catalyzed substitution of alkoxyacetate with 1,4-butyrolactone to form the keto-lactone intermediate.
- Execute selective reduction of the ketone moiety using hydride reagents like sodium borohydride or DIBAL-H.
- Complete the bicyclic structure formation via strong protic acid-catalyzed cyclization and dehydration.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthetic route offers tangible strategic benefits beyond mere technical feasibility. The elimination of exotic catalysts and hazardous reagents translates directly into a more resilient supply chain, less susceptible to disruptions caused by the scarcity of specialized raw materials. By simplifying the process flow, manufacturers can reduce the number of unit operations required, which in turn lowers capital expenditure on equipment and decreases the overall footprint of the production facility. This efficiency gain is crucial for maintaining competitive pricing in the generic pharmaceutical market, where margin pressures are intense. Furthermore, the improved safety profile reduces insurance costs and regulatory scrutiny, adding another layer of financial advantage to the organization.
- Cost Reduction in Manufacturing: The removal of expensive chiral metal catalysts and the avoidance of cryogenic cooling systems result in substantial cost savings. Traditional methods often incur high utility costs due to the energy required to maintain temperatures at -78°C, whereas this new process operates at much more moderate thermal conditions. Additionally, the use of commodity chemicals like sodium ethoxide and standard solvents reduces raw material procurement costs significantly. These factors combine to lower the total cost of goods sold, enabling more aggressive pricing strategies or improved profit margins without sacrificing product quality or regulatory compliance.
- Enhanced Supply Chain Reliability: Sourcing raw materials for this synthesis is straightforward, as the key reagents are widely produced and available from multiple vendors globally. This diversification of supply sources mitigates the risk of single-source dependency, which is a common vulnerability in pharmaceutical manufacturing. The robustness of the reaction conditions also means that production is less likely to be halted due to minor fluctuations in environmental controls or equipment performance. Consequently, this leads to more predictable delivery schedules and improved ability to meet the demanding timelines of downstream API manufacturers and finished dose formulators.
- Scalability and Environmental Compliance: The process is inherently designed for scalability, avoiding steps that are difficult to translate from laboratory to plant scale, such as photochemical reactions. The waste stream generated is less hazardous compared to nitro-based routes, simplifying effluent treatment and reducing environmental compliance costs. This alignment with green chemistry principles not only satisfies corporate sustainability goals but also future-proofs the manufacturing site against tightening environmental regulations. The ability to scale from kilograms to metric tons without fundamental process changes ensures a seamless transition from clinical supply to commercial production.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this synthesis technology. They are derived from the specific beneficial effects and background technical pain points identified in the patent documentation. Understanding these nuances is critical for R&D teams evaluating the feasibility of technology transfer and for procurement officers assessing the long-term viability of this supply source. The answers provided reflect the objective capabilities of the method as described in the intellectual property, ensuring transparency and accuracy for all stakeholders involved in the decision-making process.
Q: What are the safety advantages of this synthesis route compared to prior art?
A: Unlike previous methods requiring hazardous nitromethane or extreme cryogenic conditions at -78°C, this patented process utilizes safer solvents like methyl tert-butyl ether and operates at manageable temperatures, significantly reducing operational risk.
Q: Is this method suitable for large-scale industrial production?
A: Yes, the process avoids expensive chiral metal catalysts and photochemical steps, relying on readily available raw materials and standard unit operations, making it highly economically viable for commercial scale-up.
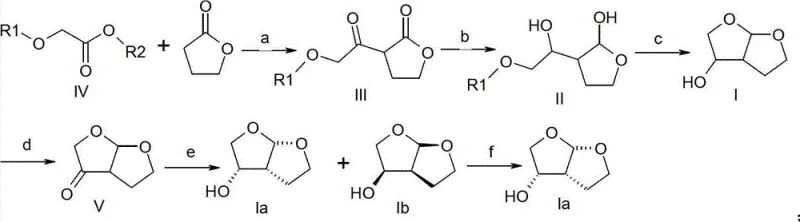
Q: What is the optical purity achievable for the enantiomer?
A: Through chiral resolution using L-di-p-toluoyl tartaric acid, the process can achieve an optical purity of approximately 96% for the target (3R,3aS,6aR) enantiomer, meeting stringent pharmaceutical specifications.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Hexahydrofuro[2,3-b]furan-3-ol Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of securing a stable and high-quality supply of key intermediates for the global fight against HIV. Our team possesses extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this patented synthesis are fully realized in practice. We operate stringent purity specifications and maintain rigorous QC labs to guarantee that every batch of hexahydrofuro[2,3-b]furan-3-ol meets the exacting standards required for API synthesis. Our commitment to technical excellence allows us to navigate the complexities of chiral resolution and cyclization with precision, delivering a product that supports the efficacy and safety of the final therapeutic agent.
We invite you to engage with our technical procurement team to discuss how this advanced synthetic route can optimize your supply chain. By requesting a Customized Cost-Saving Analysis, you can gain deeper insights into the potential economic impact of switching to this methodology. We encourage you to contact us to obtain specific COA data and route feasibility assessments tailored to your production requirements. Partnering with us ensures access to cutting-edge chemical technology backed by a reliable manufacturing infrastructure dedicated to supporting your long-term business goals.
Engineering Bottleneck?
Can't scale up this synthesis? Upload your target structure or CAS, and our CDMO team will evaluate the industrial feasibility within 24 hours. Request Evaluation →