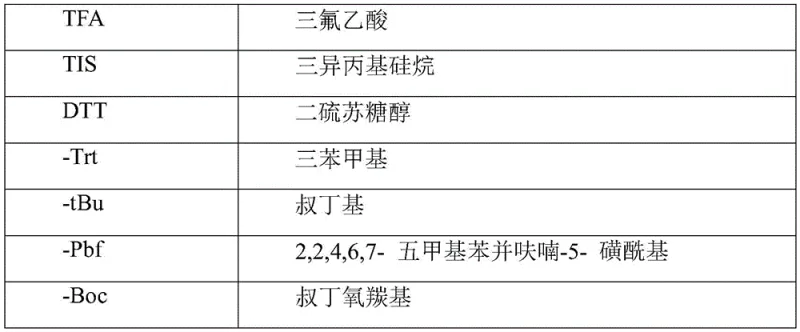
Optimizing Liraglutide Production via Novel Solid-Phase Coupling Strategies for Global API Markets
The pharmaceutical industry continuously seeks robust manufacturing routes for high-value peptide therapeutics, and the recent disclosure in patent CN110845600B offers a transformative approach to producing Liraglutide, a critical GLP-1 analogue used in diabetes management. This patent details a sophisticated solid-phase synthesis strategy that fundamentally rethinks the anchoring point of the peptide chain to the resin, specifically addressing the notorious difficulty in coupling the Glutamine (Gln) residue at the 17th position. By shifting the connection point to the side chain carboxylic acid of Glutamic Acid (Glu) linked to an amino-type resin, the inventors have created a system that enhances molecular flexibility and drastically reduces steric hindrance during the elongation process. This technical breakthrough not only resolves long-standing purity issues associated with incomplete peptide sequences but also establishes a new benchmark for efficiency in the commercial scale-up of complex peptide APIs. For procurement and R&D leaders, understanding this shift from conventional C-terminal anchoring to this novel side-chain anchoring is essential for evaluating future supply chain reliability and cost structures in the anti-diabetic market.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthesis pathways for Liraglutide, such as those described in prior patents like CN103087181 and CN102286092, typically rely on sequential Fmoc solid-phase synthesis starting from the C-terminus. These conventional methods suffer from significant drawbacks, primarily the generation of incomplete peptide sequences that possess polarity similar to the target molecule, making purification exceptionally difficult and costly. Furthermore, strategies involving the modification of the Lysine side chain after the main chain synthesis are prone to incomplete reactions or over-reaction due to high steric hindrance, leading to defective peptides and low total yields. Alternative approaches using solid-phase fragment condensation often require a massive excess of peptide fragments, ranging from 1.5 to 3.5 times the stoichiometric amount, resulting in serious material waste and inflated production costs. Additionally, methods employing liquid-phase segment condensation involve tedious post-treatment steps and struggle with solubility issues, while recombinant gene expression techniques face challenges related to high technical difficulty, substantial three-waste generation, and expensive downstream processing.
The Novel Approach
In stark contrast to these legacy methods, the novel approach disclosed in CN110845600B introduces a strategic pivot by connecting Fmoc-Glu-2-phenylisopropyl ester side chain carboxylic acid directly to an amino-type resin. This unique configuration effectively solves the problem of difficult Gln coupling by transforming the problematic Gln position into a Glu side chain attachment point that is inherently more flexible. The increased flexibility between the peptide chain and the resin allows the growing chain to deform more easily, thereby minimizing the steric hindrance that typically plagues long peptide syntheses. Moreover, this method eliminates the need for Palladium catalytic systems often used in other advanced protocols, thereby avoiding the introduction of heavy metal impurities that require complex removal steps. By synthesizing specific peptide fragments (V, VI, and VII) separately and coupling them to this central anchor, the process achieves a streamlined workflow that significantly enhances reaction efficiency and final product quality.
Mechanistic Insights into Fmoc Solid-Phase Coupling and Resin Anchoring
The core mechanistic advantage of this synthesis lies in the precise chemical engineering of the resin-peptide interface. The process begins with the coupling of Fmoc-Glu-2-R side chain carboxylic acid to amino type Resin A, utilizing robust coupling reagent combinations such as PyBOP/DIPEA/HOBt or DEPBT/DIEA. This initial step is critical as it establishes the 'middle-out' growth direction, where the peptide chain extends from the 17th position towards both the N-terminus and the C-terminus fragments. The use of phenylisopropyl ester protecting groups on the fragments ensures stability during the coupling phases while allowing for clean removal under acidic conditions later. The subsequent coupling of the pre-synthesized fragments, such as NH2-Ala-Ala-Lys(Glu(Nα-Palmitoyl)-OtBu)-phenylisopropyl ester, is facilitated by the reduced steric bulk around the reaction site, ensuring high conversion rates without the need for excessive reagent equivalents.
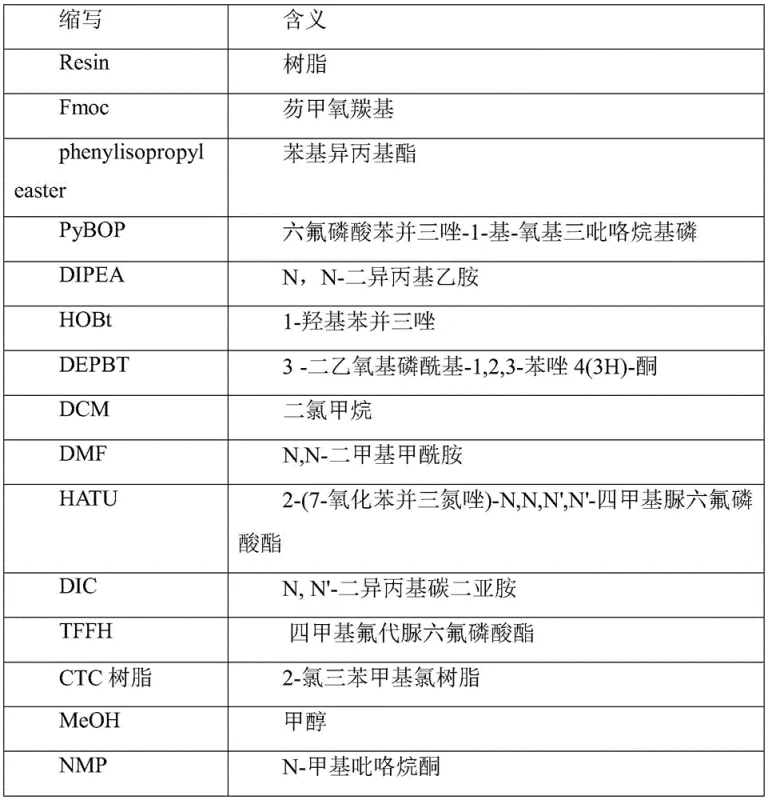
Furthermore, the impurity control mechanism is inherently built into this structural design. By avoiding the late-stage modification of the Lysine side chain on a fully assembled resin-bound peptide, the method prevents the formation of diacylation impurities and deletion sequences that are common in linear synthesis. The acidic cleavage conditions, utilizing a mixture of TFA, TIS, water, and DTT, are optimized to simultaneously remove all protecting groups and cleave the peptide from the resin without degrading the sensitive amide bonds. The result is a crude peptide with significantly higher purity (83.9% in the patent example compared to 68.1% in comparative examples), which simplifies the subsequent chromatographic purification steps. This mechanistic elegance translates directly into operational excellence, as fewer purification cycles are required to meet the stringent pharmacopeial standards for injectable peptide drugs.
How to Synthesize Liraglutide Efficiently
The synthesis of Liraglutide via this patented method involves a sequence of highly controlled solid-phase reactions that prioritize yield and purity at every stage. The process begins with the preparation of the key resin-bound intermediate, followed by the independent synthesis of protected peptide fragments which are then coupled in a specific order to build the full sequence. Detailed standard operating procedures regarding reagent concentrations, reaction times, and washing protocols are critical for replicating the high success rates reported in the patent data. For a comprehensive understanding of the specific molar ratios and temperature controls required for each coupling step, please refer to the standardized guide below.
- Connect Fmoc-Glu-2-phenylisopropyl ester side chain carboxylic acid to amino type Resin A using Fmoc solid phase synthesis to obtain the key intermediate Fmoc-Glu(Resin)-R.
- Prepare fully protected polypeptide fragments (V, VI, VII) separately on CTC resin, followed by acidolysis and carboxyl protection to form phenylisopropyl ester fragments.
- Sequentially couple the prepared peptide fragments to the resin-bound Glu intermediate, followed by final N-terminal elongation, acidic cleavage, and chromatographic purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain directors, the adoption of this synthesis methodology offers profound economic and logistical benefits that extend beyond simple yield improvements. The elimination of Palladium catalysts removes an entire unit operation dedicated to heavy metal scavenging, which not only reduces the consumption of expensive scavenging resins but also shortens the overall production cycle time. The significant increase in crude purity means that preparative HPLC columns have a longer lifespan and require less frequent regeneration, leading to substantial cost reductions in consumables and solvent usage. Furthermore, the ability to use stoichiometric or near-stoichiometric amounts of peptide fragments, rather than the 3.5-fold excess required in older fragment condensation methods, drastically lowers the raw material cost per kilogram of finished API. These efficiencies compound to create a more resilient supply chain capable of responding to market demand fluctuations without the burden of excessive inventory or waste disposal costs.
- Cost Reduction in Manufacturing: The process achieves a refined yield of 40.7%, which is nearly double the yield of comparative conventional methods that often hover around 20%. This dramatic improvement in material throughput means that for the same amount of starting amino acids and reagents, manufacturers can produce significantly more finished product, effectively halving the raw material cost contribution per gram of API. Additionally, the avoidance of Palladium catalysts eliminates the cost associated with precious metal recovery and the specialized testing required to certify low metal levels, further driving down the cost of goods sold (COGS) for the final drug substance.
- Enhanced Supply Chain Reliability: By simplifying the synthesis route and reducing the number of purification steps, the lead time for manufacturing batches is significantly compressed. The robustness of the solid-phase coupling reactions described ensures consistent batch-to-batch quality, reducing the risk of failed batches that can disrupt supply schedules. The use of commercially available reagents and standard Fmoc protection strategies means that the supply chain is not dependent on exotic or single-source custom chemicals, thereby mitigating the risk of raw material shortages and ensuring a steady flow of production for global markets.
- Scalability and Environmental Compliance: The method is explicitly designed for industrial production, with process parameters that translate seamlessly from laboratory scale to multi-ton commercial manufacturing. The reduction in solvent waste, achieved by avoiding the large excesses of fragments and minimizing purification cycles, aligns with increasingly strict environmental regulations regarding volatile organic compound (VOC) emissions. The high purity of the crude product also reduces the volume of acidic and organic waste streams generated during the polishing steps, making the facility's environmental footprint smaller and easier to manage within modern green chemistry frameworks.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this Liraglutide synthesis technology. These answers are derived directly from the experimental data and claims presented in the patent documentation, providing a factual basis for decision-making. Understanding these nuances is vital for technical teams evaluating the feasibility of technology transfer and for commercial teams assessing the competitive positioning of the resulting product in the global marketplace.
Q: How does this patent method solve the difficult coupling of Gln in Liraglutide synthesis?
A: The method connects the Glu side chain carboxylic acid to the amino resin first. This creates a longer, more flexible linkage between the peptide chain and the resin, reducing steric hindrance and allowing the difficult Gln position to couple more effectively.
Q: What are the purity and yield advantages of this novel synthesis route?
A: According to the patent data, this method achieves a refined Liraglutide yield of 40.7% with an HPLC purity of 99.85%, significantly outperforming conventional methods which often struggle with yields around 20% and lower purity due to incomplete reactions.
Q: Does this process involve heavy metal catalysts like Palladium?
A: No, unlike some prior art methods that require Palladium catalytic systems for deprotection, this novel approach avoids the use of Palladium entirely, eliminating the risk of heavy metal contamination and simplifying the purification process.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Liraglutide Supplier
At NINGBO INNO PHARMCHEM, we recognize the critical importance of adopting advanced synthesis technologies like the one described in CN110845600B to maintain competitiveness in the global peptide market. As a premier CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that the theoretical benefits of this novel route are fully realized in large-scale manufacturing. Our facilities are equipped with stringent purity specifications and rigorous QC labs capable of verifying the 99.85% HPLC purity targets set by this method, guaranteeing that every batch meets the highest international regulatory standards for safety and efficacy.
We invite potential partners to engage with our technical procurement team to discuss how this optimized synthesis route can be tailored to your specific volume requirements and cost targets. By requesting a Customized Cost-Saving Analysis, you can gain detailed insights into the potential ROI of switching to this superior manufacturing process. We encourage you to contact us today to obtain specific COA data and route feasibility assessments, ensuring that your supply chain for this vital anti-diabetic API is both economically efficient and technically robust for the long term.