Revolutionizing Semaglutide Production: Advanced Solid-Phase Synthesis for High-Purity API Intermediates
Revolutionizing Semaglutide Production: Advanced Solid-Phase Synthesis for High-Purity API Intermediates
The pharmaceutical landscape for Type 2 diabetes and obesity treatments has been fundamentally reshaped by the advent of long-acting GLP-1 analogues, with Semaglutide standing as a paramount example of modern peptide therapeutics. As detailed in patent CN112125970A, published in late 2020, a sophisticated synthetic methodology has been developed to address the notorious challenges associated with the large-scale production of this complex molecule. This patent outlines a refined solid-phase peptide synthesis (SPPS) strategy that moves beyond conventional linear assembly, introducing critical innovations in protecting group chemistry and fragment condensation. For R&D directors and procurement specialists seeking a reliable pharmaceutical intermediates supplier, understanding these technical nuances is vital, as they directly correlate to the feasibility of commercial scale-up and the consistency of the final active pharmaceutical ingredient (API). The disclosed method specifically targets the mitigation of side reactions such as racemization and beta-elimination, which have historically plagued the synthesis of long-chain peptides modified with fatty acids.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Traditional synthesis routes for Semaglutide often rely on a straightforward linear assembly of amino acids or a combination of fermentation and chemical conjugation that lacks precision. In standard Fmoc solid-phase synthesis, the accumulation of deletion sequences and truncation byproducts becomes exponential as the peptide chain lengthens beyond thirty residues. A significant bottleneck identified in prior art involves the N-terminal region, where the Histidine residue is highly prone to racemization, leading to difficult-to-separate diastereomeric impurities that compromise the safety profile of the drug. Furthermore, the hydrophobic nature of the sequence, particularly around the Val-Ser-Ser-Tyr-Leu segment, promotes the formation of rigid beta-sheet secondary structures on the resin. This aggregation causes the resin beads to shrink and become inaccessible to reagents, drastically reducing coupling efficiency and resulting in a crude product purity that often necessitates expensive and yield-loss-inducing purification steps. Additionally, the use of standard tert-butyl protection on Aspartic acid residues frequently leads to beta-elimination side reactions, generating fumarate byproducts that are structurally similar to the target molecule and extremely challenging to remove.
The Novel Approach
The methodology presented in CN112125970A offers a transformative solution by integrating orthogonal protection strategies and pre-activated peptide fragments to bypass these kinetic and thermodynamic barriers. Instead of coupling every amino acid individually, the process utilizes dipeptide and tetrapeptide building blocks, such as Fmoc-Thr(tBu)-Phe-OH and Boc-His(Trt)-Aib-Glu(OtBu)-Gly-OH, which significantly reduce the number of reaction cycles and exposure of sensitive residues to activation conditions. This fragment-based approach not only accelerates the synthesis timeline but also inherently protects the stereochemical integrity of the N-terminal Histidine and the sterically hindered Aminoisobutyric acid (Aib) at position 2. Moreover, the strategic selection of side-chain protecting groups plays a pivotal role; by substituting standard protecting groups with bulkier alternatives like Trityl (Trt) for Serine and specialized esters for Aspartic acid, the method effectively disrupts the hydrogen bonding networks that cause resin aggregation. This ensures that the growing peptide chain remains solvated and reactive throughout the synthesis, leading to a dramatic improvement in both crude purity and overall yield compared to legacy processes.
Mechanistic Insights into Specialized Protecting Group Chemistry
A cornerstone of this advanced synthesis protocol is the meticulous engineering of the Aspartic acid residue at position 9, a known hotspot for degradation via beta-elimination. In conventional peptide chemistry, the base-labile nature of the Fmoc deprotection cycle can induce the elimination of the beta-carboxyl group when protected as a simple tert-butyl ester, resulting in the formation of an alpha,beta-unsaturated aspartyl residue. To counteract this, the patent introduces the use of Fmoc-Asp(OMpe)-OH and Fmoc-Asp(OEpe)-OH, where the side chain is protected by 1-ethyl-1-methyl-propyl or 1,1-diethyl-propyl esters. These groups possess significantly greater steric hindrance compared to the tert-butyl group, physically shielding the acidic beta-proton from abstraction by the piperidine base used in Fmoc removal. This steric bulk not only suppresses the elimination pathway but also contributes to the overall solvation of the peptide chain, preventing the collapse of the resin matrix.
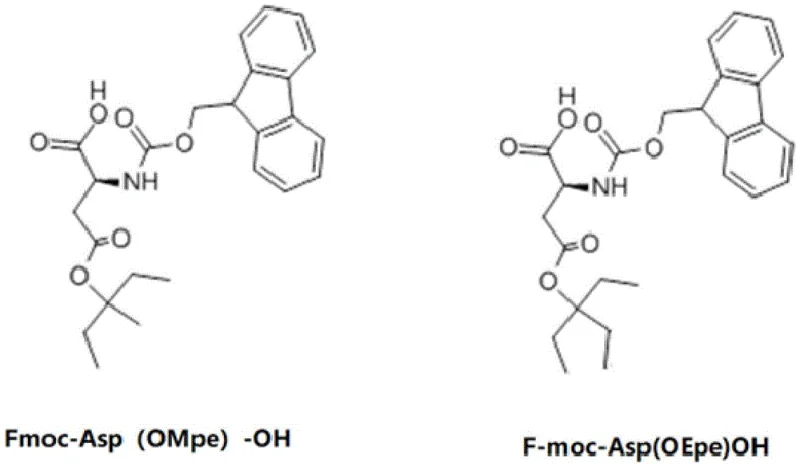
Beyond the stabilization of Aspartic acid, the method addresses the issue of beta-sheet formation through the modification of Serine residues at positions 8, 11, and 12. Standard protocols typically employ Fmoc-Ser(tBu)-OH, but the relatively small tert-butyl group fails to prevent the intermolecular hydrogen bonding between amide backbones that drives aggregation. By switching to Fmoc-Ser(Trt)-OH, the synthesis leverages the massive spatial volume of the trityl group to act as a "solubilizing tag." This bulky moiety keeps adjacent peptide chains separated on the solid support, maintaining the resin in a swollen, gel-like state that facilitates the diffusion of coupling reagents to the reactive amine termini. This mechanistic adjustment is critical for achieving high coupling yields in the hydrophobic middle section of the Semaglutide sequence, ensuring that the final crude peptide contains a minimal amount of deletion impurities. The combination of these steric and electronic modifications creates a robust synthetic environment that is far more forgiving and efficient than traditional SPPS methods.
How to Synthesize Semaglutide Efficiently
The implementation of this synthesis route requires precise control over reaction conditions and reagent stoichiometry to maximize the benefits of the specialized monomers. The process begins with the loading of Fmoc-Gly onto a suitable resin, such as Wang or 2-CTC resin, followed by the iterative addition of amino acids and fragments using potent coupling systems like HATU/HOAt or DIC/Oxyma. Critical attention must be paid to the deprotection steps, where the concentration of piperidine and the duration of treatment are optimized to remove the Fmoc group without inducing side reactions. The orthogonal deprotection of the Lysine side chain at position 20 is another key operational step, allowing for the late-stage attachment of the complex fatty acid-diacid linker. Detailed standardized operating procedures regarding temperature controls, washing protocols, and cleavage cocktails are essential for reproducibility.
- Couple Fmoc-Gly to resin, then sequentially add amino acids using Fmoc/Boc strategies, incorporating special fragments like Boc-His-Aib-Glu-Gly to prevent racemization.
- Selectively remove the Lys side-chain protecting group (Alloc, Dde, or ivDde) using specific reagents like Pd(0) or hydrazine without affecting the main chain.
- Couple the fatty acid side chain (AEEA-AEEA-gamma-Glu-Octadecanedioic acid) and cleave the peptide from the resin using TFA cocktail for final purification.
Commercial Advantages for Procurement and Supply Chain Teams
For procurement managers and supply chain heads, the technical refinements described in this patent translate directly into tangible operational benefits and risk mitigation. The primary advantage lies in the substantial reduction of downstream processing costs. By achieving a significantly higher purity in the crude peptide stage through the use of specialized protecting groups and fragment condensation, the burden on the purification department is drastically alleviated. This means less solvent consumption, reduced column chromatography time, and a higher recovery rate of the final API, all of which contribute to a lower cost of goods sold (COGS). Furthermore, the elimination of difficult-to-remove impurities, such as beta-elimination byproducts and racemized diastereomers, simplifies the quality control workflow and reduces the likelihood of batch failures, thereby enhancing the overall reliability of the supply chain.
- Cost Reduction in Manufacturing: The shift towards a more efficient synthesis route eliminates the need for excessive reagent equivalents and prolonged reaction times that are characteristic of struggling linear syntheses. By preventing resin aggregation and ensuring high coupling efficiency at every step, the process minimizes the generation of truncated sequences that would otherwise act as costly waste. The use of robust protecting groups also reduces the incidence of batch rejection due to out-of-specification impurity profiles, leading to significant economic savings over the lifecycle of the product. Additionally, the ability to use standard, commercially available coupling reagents without requiring exotic catalysts ensures that the raw material costs remain stable and predictable.
- Enhanced Supply Chain Reliability: The robustness of this synthetic method ensures a consistent supply of high-quality pharmaceutical intermediates even under fluctuating market demands. Because the process is less sensitive to minor variations in reaction conditions due to the stabilizing effects of the Trt and OMpe/OEpe groups, it offers a wider operating window for manufacturing teams. This resilience translates to shorter lead times and a more dependable delivery schedule for downstream drug manufacturers. The scalability of the solid-phase approach, combined with the high yield of the crude product, allows for seamless transition from pilot-scale batches to multi-ton commercial production without the need for extensive process re-optimization.
- Scalability and Environmental Compliance: From an environmental and regulatory standpoint, the improved efficiency of this method results in a reduced solvent footprint per kilogram of produced API. Higher yields mean less waste generation, aligning with green chemistry principles and easing the burden on waste treatment facilities. The avoidance of heavy metal catalysts in the deprotection steps (using mild hydrazine or palladium scavengers efficiently) further simplifies the compliance landscape regarding residual metals in the final drug substance. This makes the process not only economically attractive but also environmentally sustainable, a key factor for modern pharmaceutical supply chains aiming to reduce their carbon footprint.
Frequently Asked Questions (FAQ)
The following questions address common technical inquiries regarding the implementation and benefits of this advanced Semaglutide synthesis protocol. These insights are derived directly from the experimental data and mechanistic explanations provided in the patent literature, offering clarity on how this method outperforms standard industry practices. Understanding these details is crucial for technical teams evaluating potential manufacturing partners or optimizing their own internal processes for GLP-1 analogue production.
Q: How does this method prevent beta-elimination at the Asp residue?
A: The process replaces standard Fmoc-Asp(OtBu)-OH with sterically hindered variants like Fmoc-Asp(OMpe)-OH or Fmoc-Asp(OEpe)-OH, which physically block the elimination pathway and stabilize the peptide backbone.
Q: Why are dipeptide fragments used instead of single amino acids?
A: Using pre-formed fragments like Fmoc-Thr(tBu)-Phe-OH reduces the number of coupling cycles and minimizes the risk of racemization at difficult sites like the N-terminal Histidine and Aib residues.
Q: What is the advantage of using Trt protection for Serine?
A: The bulky Trityl (Trt) group on Serine prevents the formation of intermolecular hydrogen bonds that lead to beta-sheet secondary structures, thereby keeping the resin swollen and accessible for efficient coupling.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable Semaglutide Supplier
The technological breakthroughs encapsulated in patent CN112125970A represent the gold standard for modern peptide manufacturing, yet translating such complex chemistry into commercial reality requires a partner with deep technical expertise and proven infrastructure. NINGBO INNO PHARMCHEM stands at the forefront of this industry, possessing extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production. Our commitment to quality is unwavering, with stringent purity specifications enforced through rigorous QC labs equipped with state-of-the-art analytical instrumentation. We understand that the production of complex peptides like Semaglutide demands not just chemical knowledge but also precise engineering control to manage the nuances of solid-phase synthesis and purification.
We invite global pharmaceutical partners to collaborate with us to leverage these advanced synthetic routes for your next-generation therapeutic programs. By engaging with our technical procurement team, you can request a Customized Cost-Saving Analysis tailored to your specific volume requirements and quality targets. We encourage you to reach out today to obtain specific COA data and route feasibility assessments that demonstrate how our optimized processes can drive down your manufacturing costs while ensuring a secure and continuous supply of high-purity API intermediates.