Advanced Metal-Free Synthesis of 1-Alkyl-3-Aryl Indolizines for Commercial Scale-Up
Advanced Metal-Free Synthesis of 1-Alkyl-3-Aryl Indolizines for Commercial Scale-Up
The pharmaceutical industry continuously seeks robust, scalable, and cost-effective synthetic routes for nitrogen-containing heterocycles, which serve as critical scaffolds in numerous bioactive molecules. Patent CN113429407B introduces a groundbreaking advancement in this domain by disclosing a simple and efficient synthesis method for 1-alkyl-3-aryl substituted indolizine compounds. This technology represents a significant departure from conventional transition metal-catalyzed processes, utilizing instead a straightforward acid-catalyzed intramolecular dehydration strategy. The patent details two distinct yet complementary methodologies: one employing an inorganic acid aqueous solution and the other utilizing an organic acid catalyst within an organic solvent system. This dual approach ensures that manufacturers can select the optimal conditions based on substrate solubility and specific functional group tolerance, thereby maximizing yield and operational efficiency.

For R&D directors and process chemists, the significance of this patent lies in its ability to streamline the construction of the indolizine core, a structure prevalent in central nervous system depressants, anticancer agents, and antibacterial drugs. By leveraging readily available (E)-4-aryl-2-pyridyl-3-alkenyl-2-ol precursors, the invention bypasses the complex multi-step sequences often required to install the nitrogen bridge. The reaction proceeds under relatively mild thermal conditions, typically between 80°C and 130°C, and achieves high conversion rates without the need for inert atmospheres or specialized equipment. This accessibility makes the technology an attractive candidate for immediate adoption in the development of high-purity pharmaceutical intermediates, addressing the growing demand for reliable supply chains in the global API market.
The Limitations of Conventional Methods vs. The Novel Approach
The Limitations of Conventional Methods
Historically, the construction of multi-substituted indolizine compounds has relied heavily on intermolecular chemical reactions driven by transition metal complexes. Traditional protocols frequently employ expensive and potentially toxic metals such as rhodium, copper, cobalt, or palladium to facilitate cycloaddition or coupling reactions. These metal-catalyzed pathways often suffer from significant drawbacks, including the formation of difficult-to-remove metal residues that necessitate rigorous and costly purification steps to meet stringent pharmaceutical quality standards. Furthermore, alternative strategies involving pyridine ylides and alkenes via [3+2] cycloaddition followed by oxidation are often labor-intensive, requiring the preparation of unstable ylide intermediates and multiple reaction vessels. These complexities not only inflate the cost of goods but also introduce variability that can hinder consistent commercial scale-up of complex heterocycles.
The Novel Approach
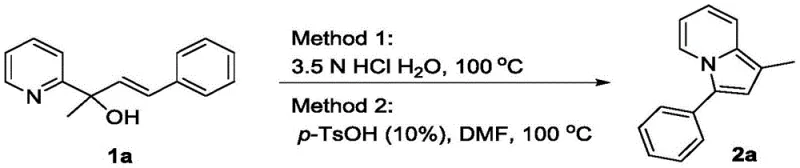
In stark contrast, the novel approach detailed in CN113429407B simplifies the synthetic landscape by utilizing a direct acid-catalyzed dehydration mechanism. As illustrated in the comparative data, the invention offers two versatile pathways that complement each other depending on the substrate nature. Method One utilizes inorganic acids like hydrochloric acid in an aqueous medium, capitalizing on the salt formation with the pyridine group to enhance solubility and drive the reaction forward. Method Two employs organic acids such as p-toluenesulfonic acid in organic solvents like DMF, providing a homogeneous environment for less water-soluble substrates. Both methods converge on the same efficient intramolecular cyclization, delivering high yields—up to 94% in optimized conditions—while completely eliminating the need for precious metal catalysts. This shift not only reduces raw material costs but also drastically simplifies the downstream processing workflow.
Mechanistic Insights into Acid-Catalyzed Intramolecular Cyclization
The core mechanistic advantage of this technology lies in the activation of the hydroxyl group and the pyridine nitrogen through protonation, facilitating a smooth intramolecular nucleophilic attack. In the inorganic acid pathway, the strong acidity of the aqueous solution protonates the pyridine nitrogen, increasing the electrophilicity of the adjacent carbon centers while simultaneously activating the hydroxyl leaving group. This dual activation lowers the energy barrier for the dehydration step, allowing the alkene chain to cyclize onto the pyridine ring efficiently. The subsequent aromatization restores the stability of the heterocyclic system, yielding the final indolizine structure. This mechanism is remarkably robust, tolerating a wide array of electronic environments on the aryl rings, which is crucial for generating diverse libraries of drug candidates.
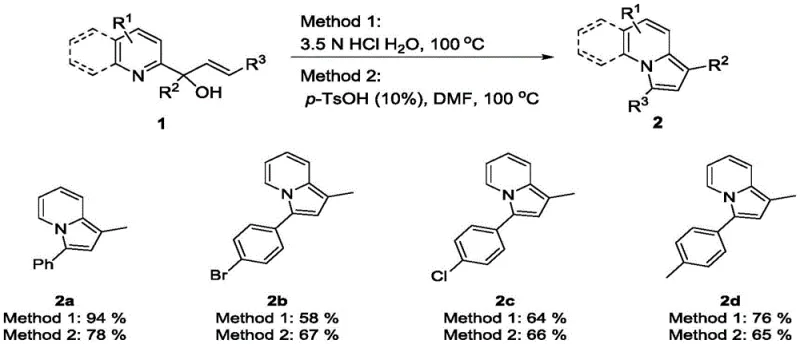
Furthermore, the substrate scope analysis reveals exceptional versatility, accommodating electron-donating groups like methoxy and methyl, as well as electron-withdrawing halogens such as bromine and chlorine. The patent data demonstrates successful synthesis across various steric environments, including bulky naphthyl and anthracenyl groups, as well as heteroaromatic systems like thiophene and benzothiophene. This broad tolerance suggests that the transition state of the cyclization is not overly sensitive to steric hindrance, likely due to the flexibility of the alkenyl linker and the driving force of aromatization. For process chemists, this means the method can be applied to a vast chemical space without extensive re-optimization, ensuring rapid progression from bench-scale discovery to pilot plant production.
How to Synthesize 1-Alkyl-3-Aryl Indolizines Efficiently
The synthesis of these valuable heterocycles is designed for operational simplicity, requiring standard laboratory or plant equipment without the need for specialized pressure vessels or gloveboxes. The process begins with the selection of the appropriate acid system based on the solubility profile of the specific (E)-4-aryl-2-pyridyl-3-alkenyl-2-ol precursor being used. Once the reaction conditions are established, the transformation proceeds via a thermal dehydration that is easy to monitor and control. For detailed procedural specifics regarding reagent ratios, temperature ramping, and workup protocols, please refer to the standardized synthesis guide below which outlines the exact steps validated in the patent examples.
- Prepare the precursor (E)-4-aryl-2-pyridyl-3-alkenyl-2-ol and select either an inorganic acid aqueous solution or an organic acid catalyst in an organic solvent.
- Heat the reaction mixture to temperatures between 80°C and 130°C under stirring for approximately 12 hours to facilitate intramolecular dehydration.
- Upon completion, neutralize the mixture if using inorganic acid, extract with ethyl acetate, and purify the crude product via silica gel column chromatography.
Commercial Advantages for Procurement and Supply Chain Teams
From a procurement and supply chain perspective, this patented methodology offers transformative benefits that directly impact the bottom line and operational reliability. By shifting away from precious metal catalysis, manufacturers can achieve substantial cost savings in raw material acquisition, as inorganic and organic acids are commodity chemicals with stable pricing and abundant global availability. This transition also mitigates the supply risk associated with fluctuating markets for rare earth metals and transition metal catalysts, ensuring a more predictable and secure production schedule for long-term contracts.
- Cost Reduction in Manufacturing: The elimination of expensive transition metal catalysts such as palladium or rhodium removes a significant cost driver from the bill of materials. Additionally, the absence of heavy metals simplifies the purification process, reducing the consumption of scavengers, specialized filtration media, and analytical testing required to verify residual metal levels. This streamlined downstream processing leads to lower operational expenditures and faster batch turnover times, enhancing overall manufacturing efficiency without compromising product quality.
- Enhanced Supply Chain Reliability: The starting materials for this synthesis, specifically the pyridyl-alkenyl-alcohol precursors, are structurally simple and can be sourced from a wide network of chemical suppliers. The robustness of the reaction conditions means that production is less susceptible to minor variations in raw material quality or environmental factors, resulting in consistent batch-to-batch reproducibility. This reliability is critical for maintaining continuous supply lines to downstream API manufacturers, reducing the risk of stockouts and delivery delays.
- Scalability and Environmental Compliance: The use of aqueous media in Method One aligns perfectly with green chemistry principles, significantly reducing the volume of volatile organic compounds (VOCs) released during production. This environmental advantage simplifies regulatory compliance and waste treatment processes, lowering the overhead costs associated with environmental health and safety management. Furthermore, the thermal nature of the reaction allows for straightforward scale-up from gram to ton quantities using standard heating and stirring infrastructure, facilitating rapid commercialization.
Frequently Asked Questions (FAQ)
The following questions address common technical and commercial inquiries regarding the implementation of this acid-catalyzed synthesis route. These insights are derived directly from the experimental data and beneficial effects described in the patent documentation, providing clarity on how this technology can be integrated into existing manufacturing workflows to optimize performance and reduce costs.
Q: What are the primary advantages of this acid-catalyzed method over traditional metal-catalyzed routes?
A: This method eliminates the need for expensive and toxic transition metal catalysts such as rhodium, copper, or palladium. It utilizes inexpensive inorganic or organic acids, significantly reducing raw material costs and simplifying the purification process by removing heavy metal residues.
Q: Does this synthesis method support a wide range of substrate substituents?
A: Yes, the patent demonstrates excellent substrate tolerance. The method successfully synthesizes indolizine derivatives with various substituents including halogens, alkyl groups, alkoxy groups, and fused aromatic systems like naphthyl and anthracenyl groups, making it highly versatile for drug discovery.
Q: Is the process suitable for large-scale industrial production?
A: Absolutely. The use of simple reagents like hydrochloric acid or p-toluenesulfonic acid, combined with standard heating and workup procedures, makes the process inherently scalable. The aqueous method particularly offers environmental benefits suitable for green manufacturing protocols.
Partnering with NINGBO INNO PHARMCHEM: Your Reliable 1-Alkyl-3-Aryl Indolizines Supplier
At NINGBO INNO PHARMCHEM, we recognize the strategic value of adopting innovative synthetic technologies like the one described in CN113429407B to enhance our service offerings. As a leading CDMO partner, we possess extensive experience scaling diverse pathways from 100 kgs to 100 MT/annual commercial production, ensuring that your transition from laboratory discovery to market supply is seamless. Our facilities are equipped with rigorous QC labs and adhere to stringent purity specifications, guaranteeing that every batch of 1-alkyl-3-aryl indolizines meets the highest international standards for pharmaceutical intermediates.
We invite you to collaborate with us to leverage this cost-effective and scalable synthesis route for your specific project needs. Our technical team is ready to provide a Customized Cost-Saving Analysis tailored to your target molecule, demonstrating exactly how this metal-free approach can optimize your budget. Please contact our technical procurement team today to request specific COA data and comprehensive route feasibility assessments, and let us help you accelerate your drug development timeline with confidence.